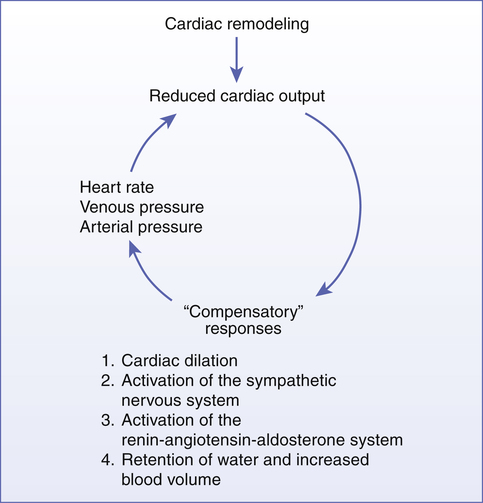
CHAPTER 48 In order to understand HF and its treatment, you need a basic understanding of hemodynamics. In particular, you need to understand the role of venous pressure, afterload, and Starling’s mechanism in determining cardiac output. You also need to understand the roles of the baroreceptor reflex, the RAAS, and the kidneys in regulating arterial pressure. If your understanding of these concepts is a little hazy, you can refresh your memory by reading Chapter 43 (Review of Hemodynamics). Because of Starling’s mechanism, the increase in heart size that occurs in HF helps improve cardiac output. That is, as the heart fails and its volume expands, contractile force increases, causing a corresponding increase in stroke volume. However, please note that the maximal contractile force that can be developed by the failing heart is considerably lower than the maximal force of the healthy heart. This limitation is reflected in the curve for the failing heart shown in Figure 48–1. • Increased heart rate. Acceleration of heart rate increases cardiac output, thereby helping improve tissue perfusion. However, if heart rate increases too much, there will be insufficient time for complete ventricular filling, and hence cardiac output will fall. • Increased contractility. Increased myocardial contractility has the obvious benefit of increasing cardiac output. The only detriment is an increase in cardiac oxygen demand. • Increased venous tone. Elevation of venous tone increases venous pressure, and thereby increases ventricular filling. Because of Starling’s mechanism, increased filling increases stroke volume. Unfortunately, if venous pressure is excessive, blood will back up behind the failing ventricles, thereby aggravating pulmonary and peripheral edema. Furthermore, excessive filling pressure can dilate the heart so much that stroke volume will begin to decline (see Fig. 48–1). • Increased arteriolar tone. Elevation of arteriolar tone increases arterial pressure, thereby increasing perfusion of vital organs. Unfortunately, increased arterial pressure also means the heart must pump against greater resistance. Since cardiac reserve is minimal in HF, the heart may be unable to meet this challenge, and output may fall. In response to stretching of the atria and dilation of the ventricles, the heart releases two natriuretic peptides: atrial natriuretic peptide (ANP) and B-natriuretic peptide (BNP). As discussed in Chapter 43, these hormones promote dilation of arterioles and veins, and also promote loss of sodium and water through the kidneys. Hence, they tend to counterbalance vasoconstriction caused by the SNS and angiotensin II, as well as retention of sodium and water caused by the RAAS. However, as HF progresses, the effects of ANP and BNP eventually become overwhelmed by the effects of the SNS and RAAS. As discussed above, reduced cardiac output leads to compensatory responses: (1) cardiac dilation, (2) activation of the SNS, (3) activation of the RAAS, and (4) retention of water and expansion of blood volume. Although these responses represent the body’s attempt to compensate for reduced cardiac output, they can actually make matters worse: Excessive heart rate can reduce ventricular filling; excessive arterial pressure can lower cardiac output; and excessive venous pressure can cause pulmonary and peripheral edema. Hence, as depicted in Figure 48–2, the “compensatory” responses can create a self-sustaining cycle of maladaptation that further impairs cardiac output and tissue perfusion. If cardiac output becomes too low to maintain sufficient production of urine, the resultant accumulation of water will eventually be fatal. The actual cause of death is complete cardiac failure secondary to excessive cardiac dilation and cardiac edema. The NYHA scheme, which has four classes, can be summarized as follows: • Class I—No limitation of ordinary physical activity • Class II—Slight limitation of physical activity: normal activity produces fatigue, dyspnea, palpitations, or angina • Class III—Marked limitation of physical activity: even mild activity produces symptoms The ACC/AHA scheme, which also has four stages, can be summarized as follows: • Stage A—At high risk for HF but without structural heart disease or symptoms of HF • Stage B—Structural heart disease but without symptoms of HF • Stage C—Structural heart disease with prior or current symptoms of HF • Stage D—Advanced structural heart disease with marked symptoms of HF at rest, and requiring specialized interventions (eg, heart transplant, mechanical assist device) Please note that the ACC/AHA scheme is intended to complement the NYHA scheme, not replace it. The relationship between the two is shown graphically in Figure 48–3. Diuretics are first-line drugs for all patients with signs of volume overload or with a history of volume overload. By reducing blood volume, these drugs can decrease venous pressure, arterial pressure (afterload), pulmonary edema, peripheral edema, and cardiac dilation. However, excessive diuresis must be avoided: If blood volume drops too low, cardiac output and blood pressure may fall precipitously, thereby further compromising tissue perfusion. For the most part, benefits of diuretics are limited to symptom reduction. As a rule, these drugs do not prolong survival. The basic pharmacology of the diuretics is discussed in Chapter 41. The RAAS plays an important role both in cardiac remodeling and in the hemodynamic changes that occur in response to reduced cardiac output. Accordingly, agents that inhibit the RAAS can be highly beneficial. Four groups of drugs are available: ACE inhibitors, ARBs, direct renin inhibitors (DRIs), and aldosterone antagonists. Of the four, the ACE inhibitors have been studied most thoroughly in HF. The basic pharmacology of the RAAS inhibitors is presented in Chapter 44. • Arteriolar dilation improves regional blood flow in the kidneys and other tissues and, by reducing afterload, it increases stroke volume and cardiac output. Increased renal blood flow promotes excretion of sodium and water. • Venous dilation reduces venous pressure, and thereby reduces pulmonary congestion, peripheral edema, preload, and cardiac dilation. • Suppression of aldosterone release enhances excretion of sodium and water, while causing retention of potassium. Adequate dosage is critical: Higher dosages are associated with increased survival. Results of the Assessment of Treatment with Lisinopril and Survival (ATLAS) trial indicate that the doses needed to increase survival are higher than those needed to produce hemodynamic changes. Unfortunately, in everyday practice, dosages are often too low: Providers frequently prescribe dosages that are large enough to produce hemodynamic benefits, but are still too low to prolong life. Target dosages associated with increased survival are summarized in Table 48–1. These dosages should be used unless side effects make them intolerable. TABLE 48–1 Inhibitors of the Renin-Angiotensin-Aldosterone System Used in Heart Failure • Promotion of myocardial remodeling (which impairs pumping) • Promotion of myocardial fibrosis (which increases the risk of dysrhythmias) • Activation of the SNS and suppression of norepinephrine uptake in the heart (both of which can promote dysrhythmias and ischemia) • Promotion of vascular fibrosis (which decreases arterial compliance) As discussed in Chapter 44, DRIs can shut down the entire RAAS. In theory, their benefits in HF should equal those of the ACE inhibitors and ARBs. At this time, only one DRI is available. This drug—aliskiren [Tekturna]—is approved for hypertension, but is not yet approved for HF. The role of beta blockers in HF continues to evolve. Until the mid-1990s, HF was considered an absolute contraindication to these drugs. After all, blockade of cardiac beta1-adrenergic receptors reduces contractility—an effect that is clearly detrimental, given that contractility is already compromised in the failing heart. However, it is now clear that, with careful control of dosage, beta blockers can improve patient status. Controlled trials have shown that three beta blockers—carvedilol [Coreg], bisoprolol [Zebeta], and sustained-release metoprolol [Toprol XL]—when added to conventional therapy, can improve LV ejection fraction, increase exercise tolerance, slow progression of HF, reduce the need for hospitalization, and, most importantly, prolong survival. Accordingly, beta blockers are now recommended for most patients. These drugs can even be used in patients with severe disease (NYHA Class IV), provided the patient is euvolemic and hemodynamically stable. Although the mechanism underlying benefits is uncertain, likely possibilities include protecting the heart from excessive sympathetic stimulation and protecting against dysrhythmias. Because excessive beta blockade can reduce contractility, doses must be very low initially and then gradually increased. Full benefits may not be seen for 1 to 3 months. Among patients with HF, the principal adverse effects are (1) fluid retention and worsening of HF, (2) fatigue, (3) hypotension, and (4) bradycardia or heart block. The basic pharmacology of the beta blockers is discussed in Chapter 18. Inotropic agents (other than digoxin) Sympathomimetic drugs: dopamine and dobutamine The basic pharmacology of dopamine and dobutamine is presented in Chapter 17. Discussion here is limited to their use in HF. Both drugs are administered by IV infusion. Vasodilators (other than ACE inhibitors and ARBs) Isosorbide dinitrate plus hydralazine Isosorbide dinitrate [Isordil, others] belongs to the same family as nitroglycerin. Like nitroglycerin, ISDN causes selective dilation of veins. In patients with severe, refractory HF, the drug can reduce congestive symptoms and improve exercise capacity. In addition to its hemodynamic actions, ISDN may inhibit abnormal myocyte growth, and hence may retard cardiac remodeling. Principal adverse effects are orthostatic hypotension and reflex tachycardia. The basic pharmacology of ISDN and other organic nitrates is discussed in Chapter 51 (Drugs for Angina Pectoris). Hydralazine [Apresoline] causes selective dilation of arterioles. By doing so, the drug can improve cardiac output and renal blood flow. For treatment of HF, hydralazine is always used in combination with ISDN, since hydralazine by itself is not very effective. Principal adverse effects are hypotension, tachycardia, and a syndrome that resembles systemic lupus erythematosus. The basic pharmacology of hydralazine is discussed in Chapter 46 (Vasodilators). In 2005, the Food and Drug Administration approved BiDil, a fixed-dose combination of hydralazine and isosorbide dinitrate, for treating HF—but only in African Americans, making BiDil the first medication approved for a specific ethnic group. Can BiDil help people in other ethnic groups? Probably, but data are lacking: The manufacturer only tested the product in blacks. As discussed in Chapter 8 (under the heading Race), testing was limited to blacks primarily because of regulatory and market incentives, not because there were data suggesting it wouldn’t work for others. Of course, now that BiDil is approved, clinicians may prescribe it for anyone they see fit. Each BiDil tablet contains 37.5 mg hydralazine and 20 mg isosorbide dinitrate. The recommended dosage is 1 or 2 tablets 3 times a day.
Drugs for heart failure
Pathophysiology of heart failure
Physiologic adaptations to reduced cardiac output
Cardiac dilation.
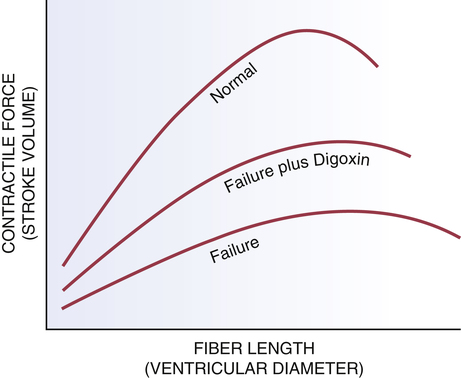
 Relationship of ventricular diameter to contractile force.
Relationship of ventricular diameter to contractile force.
In the normal heart and the failing heart, increased fiber length produces increased contractile force. However, for any given fiber length, contractile force in the failing heart is much less than in the healthy heart. By increasing cardiac contractility, digoxin shifts the relationship between fiber length and stroke volume in the failing heart toward that in the normal heart.
Increased sympathetic tone.
Natriuretic peptides.
The vicious cycle of “compensatory” physiologic responses
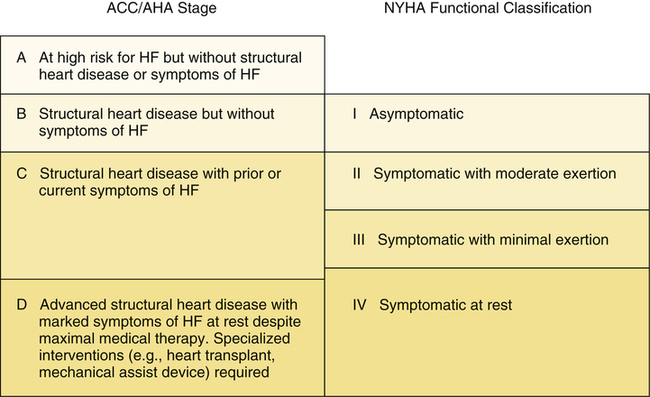
Classification of heart failure severity
Overview of drugs used to treat heart failure
Diuretics
Drugs that inhibit the RAAS
ACE inhibitors
Hemodynamic benefits.
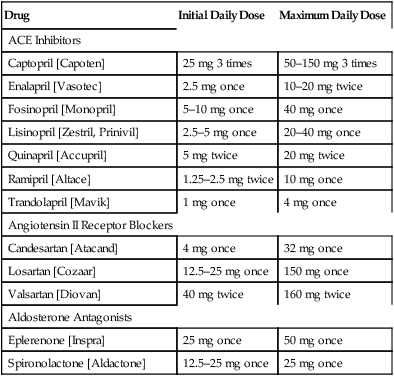
Dosage.

Drug
Initial Daily Dose
Maximum Daily Dose
ACE Inhibitors
Captopril [Capoten]
25 mg 3 times
50–150 mg 3 times
Enalapril [Vasotec]
2.5 mg once
10–20 mg twice
Fosinopril [Monopril]
5–10 mg once
40 mg once
Lisinopril [Zestril, Prinivil]
2.5–5 mg once
20–40 mg once
Quinapril [Accupril]
5 mg twice
20 mg twice
Ramipril [Altace]
1.25–2.5 mg twice
10 mg once
Trandolapril [Mavik]
1 mg once
4 mg once
Angiotensin II Receptor Blockers
Candesartan [Atacand]
4 mg once
32 mg once
Losartan [Cozaar]
12.5–25 mg once
150 mg once
Valsartan [Diovan]
40 mg twice
160 mg twice
Aldosterone Antagonists
Eplerenone [Inspra]
25 mg once
50 mg once
Spironolactone [Aldactone]
12.5–25 mg once
25 mg once
Aldosterone antagonists
Direct renin inhibitors
Beta blockers
Digoxin
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree



Drugs for heart failure


Get Clinical Tree app for offline access