CHAPTER 44 Before considering the physiology of the RAAS, we need to introduce the angiotensin family, which consists of angiotensin I, angiotensin II, and angiotensin III. All three compounds are small polypeptides. Angiotensin I is the precursor of angiotensin II (Fig. 44–1) and has only weak biologic activity. In contrast, angiotensin II has strong biologic activity. And angiotensin III, which is formed by degradation of angiotensin II, has moderate biologic activity. As indicated in Figure 44–1, angiotensin II is formed through two sequential reactions. The first is catalyzed by renin, the second by ACE. As indicated in Figure 44–1, release of renin can be triggered by multiple factors. Release increases in response to a decline in blood pressure, blood volume, plasma sodium content, or renal perfusion pressure. Reduced renal perfusion pressure is an especially important stimulus for renin release, and can occur in response to (1) stenosis of the renal arteries, (2) reduced systemic blood pressure, and (3) reduced plasma volume (brought on by dehydration, hemorrhage, or chronic sodium depletion). For the most part, these factors increase renin release through effects exerted locally in the kidney. However, some of these factors may also promote renin release through activation of the sympathetic nervous system. (Sympathetic nerves increase secretion of renin by causing stimulation of beta1-adrenergic receptors on juxtaglomerular cells.) As depicted in Figure 44–1, the RAAS, acting through angiotensin II, raises blood pressure through two basic processes: vasoconstriction and renal retention of water and sodium. Vasoconstriction raises blood pressure by increasing total peripheral resistance; retention of water and sodium raises blood pressure by increasing blood volume. Vasoconstriction occurs within minutes to hours of activating the system, and hence can raise blood pressure quickly. In contrast, days, weeks, or even months are required for the kidney to raise blood pressure by increasing blood volume. As suggested by Figure 44–1, angiotensin II acts in two ways to promote renal retention of water. First, by constricting renal blood vessels, angiotensin II reduces renal blood flow, and thereby reduces glomerular filtration. Second, angiotensin II stimulates release of aldosterone from the adrenal cortex. Aldosterone then acts on renal tubules to promote retention of sodium and water and excretion of potassium. As indicated in Figure 44–2, ACE inhibitors produce their beneficial effects and adverse effects by (1) reducing levels of angiotensin II (through inhibition of ACE) and (2) increasing levels of bradykinin (through inhibition of kinase II). By reducing levels of angiotensin II, ACE inhibitors can dilate blood vessels (primarily arterioles and to a lesser extent veins), reduce blood volume (through effects on the kidney), and, importantly, prevent or reverse pathologic changes in the heart and blood vessels mediated by angiotensin II and aldosterone. Inhibition of ACE can also cause hyperkalemia and fetal injury. Elevation of bradykinin causes vasodilation (secondary to increased production of prostaglandins and nitric oxide), and can also promote cough and angioedema. Regarding pharmacokinetics, the following generalizations apply: • Nearly all ACE inhibitors are administered orally. The only exception is enalaprilat (the active form of enalapril), which is given IV. • Except for captopril and moexipril, all oral ACE inhibitors can be administered with food. • With the exception of captopril, all ACE inhibitors have prolonged half-lives, and hence can be administered just once or twice a day. Captopril is administered 2 or 3 times a day. • With the exception of lisinopril, all ACE inhibitors are prodrugs that must undergo conversion to their active form in the small intestine and liver. Lisinopril is active as given. • All ACE inhibitors are excreted by the kidneys. As a result, nearly all can accumulate to dangerous levels in patients with kidney disease, and hence dosages must be reduced in these patients. Only one agent—fosinopril—does not require a dosage reduction. When the ACE inhibitors were introduced (over 30 years ago), their only indication was hypertension. Today, they are also used for heart failure, acute MI, left ventricular dysfunction, and diabetic and nondiabetic nephropathy. In addition, they can help prevent MI, stroke, and death in patients at high risk for cardiovascular events. It should be noted that no single ACE inhibitor is approved for all of these conditions (Table 44–1). However, given that all ACE inhibitors are very similar, it seems likely that all may produce similar benefits. TABLE 44–1 ACE Inhibitors: Approved Indications and Adult Dosages *CAD = coronary artery disease, CVD = cardiovascular disease, LVD = left ventricular dysfunction, MI = myocardial infarction. †For all ACE inhibitors except fosinopril, dosage must be reduced in patients with significant renal impairment. ACE inhibitors offer several advantages over most other antihypertensive drugs. In contrast to the sympatholytic agents, ACE inhibitors do not interfere with cardiovascular reflexes. Hence, exercise capacity is not impaired and orthostatic hypotension is minimal. In addition, these drugs can be used safely in patients with bronchial asthma, a condition that precludes the use of beta2-adrenergic antagonists. ACE inhibitors do not promote hypokalemia, hyperuricemia, or hyperglycemia—side effects seen with thiazide diuretics. Furthermore, they do not induce lethargy, weakness, or sexual dysfunction—responses that are common with other antihypertensive agents. Most importantly, ACE inhibitors reduce the risk of cardiovascular mortality caused by hypertension. The only other drugs proved to reduce hypertension-associated mortality are beta blockers and diuretics (see Chapter 47). ACE inhibitors produce multiple benefits in heart failure. By lowering arteriolar tone, these drugs improve regional blood flow, and, by reducing cardiac afterload, they increase cardiac output. By causing venous dilation, they reduce pulmonary congestion and peripheral edema. By dilating blood vessels in the kidney, they increase renal blood flow, and thereby promote excretion of sodium and water. This loss of fluid has two beneficial effects: (1) it helps reduce edema and (2) by lowering blood volume, it decreases venous return to the heart, and thereby reduces right-heart size. Lastly, by suppressing aldosterone and reducing local production of angiotensin II in the heart, ACE inhibitors may prevent or reverse pathologic changes in cardiac structure. Although only seven ACE inhibitors are approved for heart failure (see Table 44–1), both the American Heart Association and the American College of Cardiology have concluded that the ability to improve symptoms and prolong survival is a class effect. The use of ACE inhibitors in heart failure is discussed further in Chapter 48. The principal protective mechanism appears to be reduction of glomerular filtration pressure. ACE inhibitors lower filtration pressure by reducing levels of angiotensin II, a compound that can raise filtration pressure by two mechanisms. First, angiotensin II raises systemic blood pressure, which raises pressure in the afferent arteriole of the glomerulus (Fig. 44–3). Second, it constricts the efferent arteriole, thereby generating back-pressure in the glomerulus. The resultant increase in filtration pressure promotes injury. By reducing levels of angiotensin II, ACE inhibitors lower glomerular filtration pressure, and thereby slow development of renal injury.
Drugs acting on the Renin-Angiotensin-Aldosterone system
Physiology of the Renin-Angiotensin-Aldosterone system
Types of angiotensin
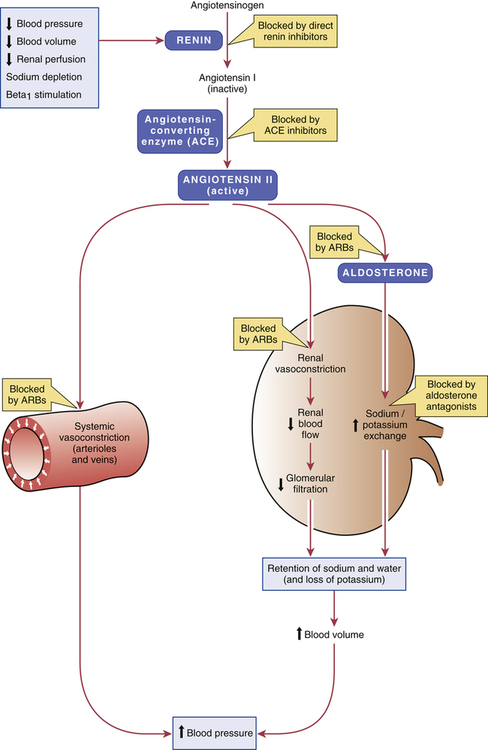
 Regulation of blood pressure by the renin-angiotensin-aldosterone system.
Regulation of blood pressure by the renin-angiotensin-aldosterone system.
In addition to the mechanisms depicted, angiotensin II can raise blood pressure by (1) acting on the distal nephron to promote reabsorption of sodium and (2) increasing vasoconstriction by three mechanisms: promoting release of norepinephrine from sympathetic nerves; promoting release of epinephrine from the adrenal medulla; and acting in the central nervous system to increase sympathetic outflow to blood vessels. (ARBs = angiotensin receptor blockers.)
Formation of angiotensin II by renin and angiotensin-converting enzyme
Renin
Regulation of renin release.
Regulation of blood pressure by the Renin-Angiotensin-Aldosterone system
Angiotensin-converting enzyme inhibitors
Mechanism of action and overview of pharmacologic effects
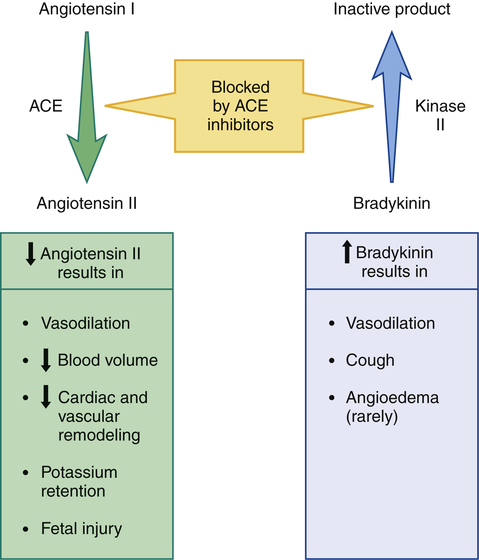
 Overview of ACE inhibitor actions and pharmacologic effects.
Overview of ACE inhibitor actions and pharmacologic effects.
Angiotensin-converting enzyme (ACE) and kinase II are two names for the same enzyme. When angiotensin II is the substrate, we call the enzyme ACE; when bradykinin is the substrate, we call it kinase II. Inhibition of this enzyme decreases production of angiotensin II (thereby reducing angiotensin II levels), and decreases breakdown of bradykinin (thereby increasing bradykinin levels).
Pharmacokinetics
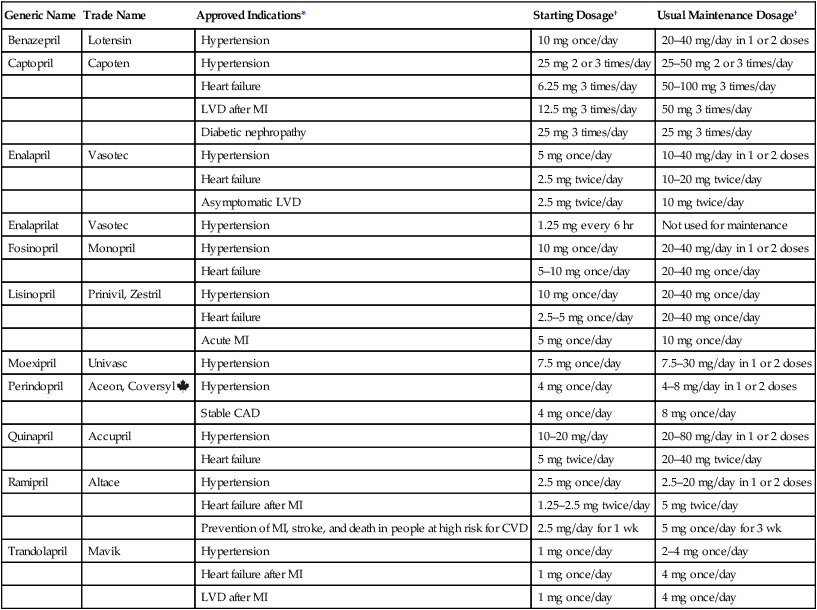
Therapeutic uses

Generic Name
Trade Name
Approved Indications*
Starting Dosage†
Usual Maintenance Dosage†
Benazepril
Lotensin
Hypertension
10 mg once/day
20–40 mg/day in 1 or 2 doses
Captopril
Capoten
Hypertension
25 mg 2 or 3 times/day
25–50 mg 2 or 3 times/day
Heart failure
6.25 mg 3 times/day
50–100 mg 3 times/day
LVD after MI
12.5 mg 3 times/day
50 mg 3 times/day
Diabetic nephropathy
25 mg 3 times/day
25 mg 3 times/day
Enalapril
Vasotec
Hypertension
5 mg once/day
10–40 mg/day in 1 or 2 doses
Heart failure
2.5 mg twice/day
10–20 mg twice/day
Asymptomatic LVD
2.5 mg twice/day
10 mg twice/day
Enalaprilat
Vasotec
Hypertension
1.25 mg every 6 hr
Not used for maintenance
Fosinopril
Monopril
Hypertension
10 mg once/day
20–40 mg/day in 1 or 2 doses
Heart failure
5–10 mg once/day
20–40 mg once/day
Lisinopril
Prinivil, Zestril
Hypertension
10 mg once/day
20–40 mg once/day
Heart failure
2.5–5 mg once/day
20–40 mg once/day
Acute MI
5 mg once/day
10 mg once/day
Moexipril
Univasc
Hypertension
7.5 mg once/day
7.5–30 mg/day in 1 or 2 doses
Perindopril
Aceon, Coversyl 
Hypertension
4 mg once/day
4–8 mg/day in 1 or 2 doses
Stable CAD
4 mg once/day
8 mg once/day
Quinapril
Accupril
Hypertension
10–20 mg/day
20–80 mg/day in 1 or 2 doses
Heart failure
5 mg twice/day
20–40 mg twice/day
Ramipril
Altace
Hypertension
2.5 mg once/day
2.5–20 mg/day in 1 or 2 doses
Heart failure after MI
1.25–2.5 mg twice/day
5 mg twice/day
Prevention of MI, stroke, and death in people at high risk for CVD
2.5 mg/day for 1 wk
5 mg once/day for 3 wk
Trandolapril
Mavik
Hypertension
1 mg once/day
2–4 mg once/day
Heart failure after MI
1 mg once/day
4 mg once/day
LVD after MI
1 mg once/day
4 mg once/day
Hypertension.
Heart failure.
Diabetic and nondiabetic nephropathy.
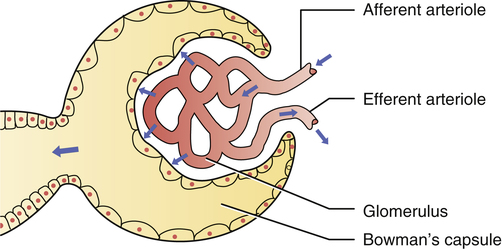
 Elevation of glomerular filtration pressure by angiotensin II.
Elevation of glomerular filtration pressure by angiotensin II.
Angiotensin II increases filtration pressure by (1) increasing pressure in the afferent arteriole (secondary to increasing systemic arterial pressure), and by (2) constricting the efferent arteriole, thereby generating back-pressure in the glomerulus.
< div class='tao-gold-member'>
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Drugs acting on the Renin-Angiotensin-Aldosterone system
 ] can reduce morbidity and mortality in patients at risk for major cardiovascular events. However, the drug is not yet approved for this use. Benefits were demonstrated in the EURopean trial On reduction of cardiac events with Perindopril in stable coronary Artery disease (EUROPA). Patients in EUROPA were at lower risk than those in HOPE.
] can reduce morbidity and mortality in patients at risk for major cardiovascular events. However, the drug is not yet approved for this use. Benefits were demonstrated in the EURopean trial On reduction of cardiac events with Perindopril in stable coronary Artery disease (EUROPA). Patients in EUROPA were at lower risk than those in HOPE.Only gold members can continue reading. Log In or Register to continue
Get Clinical Tree app for offline access
