*All dose modifications should be based on the worst preceding toxicity.
†Common Terminology Criteria for Adverse Events.
Onivyde
There is no recommended dose of ONIVYDE for patients with serum bilirubin above the upper limit of normal. ■ Withhold ONIVYDE for an absolute neutrophil count below 1,500/mm3 or neutropenic fever. ■ Withhold ONIVYDE for diarrhea of Grade 2 to 4 severity. ■ See the following chart for dose modifications recommended for adverse reactions.
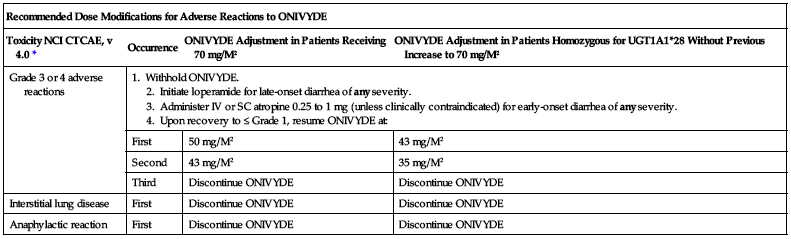
| Recommended Dose Modifications for Adverse Reactions to ONIVYDE | |||
| Toxicity NCI CTCAE, v 4.0* | Occurrence | ONIVYDE Adjustment in Patients Receiving 70 mg/M2 | ONIVYDE Adjustment in Patients Homozygous for UGT1A1*28 Without Previous Increase to 70 mg/M2 |
| Grade 3 or 4 adverse reactions | 1. Withhold ONIVYDE. 2. Initiate loperamide for late-onset diarrhea of any severity. 3. Administer IV or SC atropine 0.25 to 1 mg (unless clinically contraindicated) for early-onset diarrhea of any severity. 4. Upon recovery to ≤ Grade 1, resume ONIVYDE at: | ||
| First | 50 mg/M2 | 43 mg/M2 | |
| Second | 43 mg/M2 | 35 mg/M2 | |
| Third | Discontinue ONIVYDE | Discontinue ONIVYDE | |
| Interstitial lung disease | First | Discontinue ONIVYDE | Discontinue ONIVYDE |
| Anaphylactic reaction | First | Discontinue ONIVYDE | Discontinue ONIVYDE |
*NCI CTCAE, v 4.0, National Cancer Institute Common Toxicity Criteria for Adverse Events, version 4.0.
For recommended dose modifications for fluorouracil (5-FU) and/or leucovorin (LV), refer to their monographs and/or full prescribing information.
Dilution
Specific techniques required; see Precautions.
Conventional irinotecan:
Available in single-dose vials in 2-mL, 5-mL, and 15-mL sizes with a concentration of 20 mg/mL. Must be diluted for infusion with D5W (preferred) or NS to concentrations between 0.12 and 2.8 mg/mL. Usually diluted in 250 to 500 mL D5W.
Onivyde:
Available in a single-dose vial containing 43 mg irinotecan free base at a concentration of 4.3 mg/mL. It is a slightly yellow, opaque, liposomal dispersion. Withdraw the calculated volume from the vial and dilute in 500 mL D5W or NS. Mix by gentle inversion. Protect diluted solution from light.
Filters:
Conventional irinotecan:
Specific information not available. ONIVYDE: Do not use in-line filters.
Storage:
Conventional irinotecan:
Packaged in a blister pack to protect against accidental breakage and leakage. Store in carton protected from light at CRT. When mixed with D5W, is chemically and physically stable for 48 hours if refrigerated and in ambient fluorescent lighting and for 24 hours at CRT. Do not refrigerate if mixed with NS; a precipitate may form. Stable for 24 hours at CRT when mixed in NS. Because of the risk of microbial contamination, the manufacturer recommends that solutions mixed in D5W or NS be used within 4 hours if kept at RT. However, if reconstitution and dilution are performed under strict aseptic conditions (e.g., on a laminar air flow bench), the solution should be used (i.e., infusion completed) within 12 hours at RT or 24 hours (D5W) if refrigerated. Avoid freezing.
Onivyde:
Refrigerate in carton at 2° to 8° C (36° to 46° F) to protect from light. Do not freeze. Administer diluted solution within 4 hours of preparation when stored at RT or within 24 hours if refrigerated. Allow diluted solution to come to RT before administration. Do not freeze. Discard unused portion.
Compatibility (underline indicates conflicting compatibility information)
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
Conventional irinotecan
Manufacturer states, “Other drugs should not be added to the infusion solution.” Consider specific use and toxicity.
One source suggests the following compatibilities:
Y-site:
Oxaliplatin (Eloxatin) and palonosetron (Aloxi).
Onivyde
Specific information not available. Consider specific use and toxicity; consult pharmacist.
Rate of administration
Both formulations:
A single dose as an infusion equally distributed over 90 minutes.
Onivyde:
Do not use in-line filter.
Actions
Conventional irinotecan:
A semi-synthetic derivative of camptothecin. An alkaloid extract from plants such as Camptotheca acuminata. A class of antineoplastic agent that inhibits the enzyme topoisomerase I required for DNA replication. Together with its active metabolite SN-38 it causes cell death by damaging DNA produced during the S-phase of cell synthesis. Maximum plasma SN-38 levels are reached within 1 hour of infusion end. Extensively distributed to body tissues. Terminal half-life of irinotecan is about 6 to 12 hours; SN-38 is 10 to 20 hours. Irinotecan is moderately bound to plasma proteins (30% to 68%), but SN-38 is highly bound (95%). Metabolic conversion of irinotecan to SN-38 primarily occurs in the liver. SN-38 is conjugated to a glucuronide metabolite by the enzyme UDP-glucuronosyltransferase 1A1 (UGT1A1). Approximately 25% to 50% excreted through bile and urine.
Onivyde:
A topoisomerase 1 inhibitor encapsulated in a lipid bilayer vesicle or liposome. Topoisomerase 1 relieves torsional strain in DNA by inducing single-strand breaks. Irinotecan and its active metabolite SN-38 bind reversibly to the topoisomerase 1–DNA complex and prevent re-ligation of the single-strand breaks, leading to cell death. 95% of irinotecan remains liposome-encapsulated. Terminal elimination half-life is approximately 25.8 hours. Protein binding is less than 0.44% of the total irinotecan in ONIVYDE. Metabolism and excretion of irinotecan liposome have not been evaluated.
Indications and uses
Conventional irinotecan:
As a component of first-line therapy in combination with 5-fluorouracil (5-FU) and leucovorin (LV) for patients with metastatic carcinoma of the colon or rectum. ■ For patients with metastatic carcinoma of the colon or rectum whose disease has recurred or progressed following initial fluorouracil-based therapy.
Onivyde:
Treatment of patients with metastatic adenocarcinoma of the pancreas after disease progression following gemcitabine-based therapy. Used in combination with fluorouracil and leucovorin.
Limitation of use of onivyde:
Not indicated as a single agent for the treatment of patients with metastatic adenocarcinoma of the pancreas.
Contraindications
Both formulations:
History of hypersensitivity to conventional or liposomal formulations of irinotecan or any of their components.
Precautions
Both formulations:
Follow guidelines for handling cytotoxic agents. See Appendix A, p. 1331. ■ Administered by or under the direction of the physician specialist. ■ Adequate diagnostic and treatment facilities must be available. ■ Can cause severe or life-threatening neutropenia and fatal neutropenic sepsis. ■ Patients who are homozygous for the UGT1A1*28 allele have decreased UGT1A1 enzyme activity. This leads to a higher exposure to SN-38 and an increased risk for neutropenia; see Dose Adjustments. A laboratory test is available to determine the UGT1A1 status of patients. ■ Can cause severe or life-threatening diarrhea that may be either early or late onset (see discussion under individual agents). ■ Severe hypersensitivity reactions, including anaphylaxis, have been reported. ■ Interstitial pulmonary disease has been reported and can be fatal. Use caution in patients with pleural effusions and/or impaired pulmonary function. Risk factors may include pre-existing lung disease or use of pneumotoxic agents (e.g., amiodarone [Nexterone]), radiation therapy, or colony-stimulating factors; see Antidote.
Conventional irinotecan:
Can induce both early and late forms of diarrhea that appear to be mediated by different mechanisms. Both forms may be severe. Interrupt therapy and reduce subsequent doses if severe diarrhea occurs; see Dose Adjustments. Late diarrhea may be complicated by colitis, ulceration, bleeding, ileus, obstruction, and infection. Cases of megacolon and intestinal perforation have been reported. Patients must not be treated with irinotecan until resolution of any bowel obstruction. ■ Hepatic dysfunction may impair the metabolism of both irinotecan and SN-38. Patients with a bilirubin of 1 to 2 mg/dL are at increased risk for developing Grade 3 or 4 neutropenia. The manufacturer does not recommend a dose for patients with a bilirubin greater than 2 mg/dL and states that insufficient information is available. ■ May cause severe myelosuppression. Deaths due to sepsis following severe neutropenia have been reported. ■ Use with caution in patients with renal impairment. Has not been studied. Not recommended for use in patients on dialysis. ■ Use caution in the elderly (may have an increased incidence and severity of diarrhea) and in patients who have had previous cytotoxic therapy or previous pelvic/abdominal irradiation (likely to have an increased incidence and severity of myelosuppression). Monitor closely. ■ Use caution in patients with poor performance status. Patients with a baseline performance status of 2 had higher rates of hospitalization, neutropenic fever, thromboembolism, first-cycle treatment discontinuation, and early deaths than patients with a performance status of 0 or 1. ■ Contains sorbitol. Do not use in patients with hereditary fructose intolerance. ■ The Mayo Clinic regimen of 5-FU/LV (administered for 4 to 5 days every 28 days) should not be used in combination with irinotecan outside a carefully controlled, well-designed clinical study. Regimen has caused increased toxicity and death. ■ See Monitor.
Onivyde:
Severe or life-threatening neutropenia, neutropenic fever or sepsis, and fatal neutropenic sepsis have occurred. The incidence of Grade 3 or 4 neutropenia was higher among Asian patients compared with Caucasian patients in clinical trials. Withhold ONIVYDE for absolute neutrophil count below 1,500/mm3 or neutropenic fever; see Monitor. ■ Severe diarrhea has occurred in patients receiving liposomal irinotecan in combination with fluorouracil and leucovorin. Withhold ONIVYDE for diarrhea of Grade 2 to 4 severity. An individual patient may experience both early- and late-onset diarrhea; see Monitor. ■ Do not administer ONIVYDE to patients with bowel obstruction. ■ Not studied in patients with hepatic impairment. ■ No pharmacokinetic effects noted in patients with mild to moderate renal impairment. Data for severe renal impairment are insufficient.
Monitor:
Both formulations:
Prophylactic antiemetics are recommended; see Usual Dose. To reduce nausea and vomiting and increase patient comfort after initial dosing, additional antiemetics should be available (e.g., prochlorperazine [Compazine], ondansetron [Zofran], granisetron [Kytril]). ■ Monitor vital signs. ■ Obtain an accurate bowel history to evaluate changes in bowel habits after administration of irinotecan. ■ Monitor for “early” diarrhea. Occurs during or within 24 hours of irinotecan administration and is cholinergic in nature. Usually transient and only infrequently is severe. May be accompanied by other cholinergic symptoms (e.g., abdominal cramping, bradycardia, diaphoresis, flushing, increased salivation, lacrimation, miosis, rhinitis). Patients who have a cholinergic reaction to irinotecan will probably have similar reactions to subsequent doses. Atropine 0.25 to 1 mg IV or SC may be considered for treatment or for prophylactic use unless clinically contraindicated. ■ Monitor for “late” diarrhea (more than 24 hours after irinotecan administration), which probably results from cytotoxic effects on GI epithelium. May be prolonged; may cause dehydration, electrolyte imbalances, or sepsis; and can be life threatening. At first onset, give loperamide (Imodium) 4 mg; give 2 mg every 2 hours (4 mg every 4 hours during the night) until diarrhea-free for a minimum of 12 hours. Monitor carefully; replace fluids and electrolytes as needed; see Patient Education. ■ Maintain adequate hydration. Orthostatic hypotension or dizziness may indicate dehydration. ■ Initiate antibiotic therapy in patients who develop ileus, fever, or severe neutropenia. ■ Be alert for signs of bone marrow suppression or infection. Prophylactic antibiotics may be indicated pending results of C/S in a febrile or nonfebrile neutropenic patient. ■ Monitor for thrombocytopenia (platelet count less than 50,000/mm3). Initiate precautions to prevent excessive bleeding (e.g., inspect IV sites, skin, and mucous membranes; use extreme care during invasive procedures; test urine, emesis, stool, and secretions for occult blood). ■ Monitor for S/S of a hypersensitivity reaction (e.g., chest pain, chills, dizziness, dyspnea, fever, flushing, hypotension, nausea, pruritus, rash, urticaria) or an infusion reaction (e.g., asthenia, chills, fatigue, fever, vomiting). ■ Monitor for S/S of interstitial lung disease. Withhold therapy in patients with new or progressive cough, dyspnea, and fever pending diagnostic evaluation; see Precautions and Antidote. ■ See Dose Adjustments.
Conventional irinotecan:
Obtain a WBC with differential, hemoglobin, and platelet count before each dose. Expected nadir for platelets is 14 days, and 21 days for hemoglobin, neutrophils, and leukocytes. ■ Obtain baseline electrolytes and liver function tests. ■ Not a vesicant, but monitor injection site for inflammation and/or extravasation. ■ If late-onset diarrhea develops, subsequent weekly chemotherapy treatments should be delayed until pretreatment bowel function has returned for at least 24 hours without the need for antidiarrheal medication. If Grade 2, 3, or 4 late diarrhea recurs, subsequent doses of irinotecan should be decreased; see Dose Adjustments. ■ Avoid use of diuretics and laxatives in patients with diarrhea. ■ Monitor renal function and hydration status. Rare cases of renal impairment or acute renal failure have been reported, usually in patients who became dehydrated from vomiting and/or diarrhea.
Onivyde (liposomal irinotecan):
Prophylactic antiemetics and corticosteroids are recommended 30 minutes before ONIVYDE infusion. ■ Monitor CBC on Days 1 and 8 of every cycle and more frequently if clinically indicated. Withhold ONIVYDE if the absolute neutrophil count (ANC) is below 1,500/mm3 or if neutropenic fever occurs. Resume when the ANC is 1,500 mm3 or above; see Dose Adjustments. ■ Monitor for infusion reactions occurring on the day of administration. S/S have included periorbital edema, pruritus, rash, and urticaria.
Patient education:
Both formulations:
Review manufacturer’s medication guide. ■ Report any unusual or unexpected symptoms or side effects as soon as possible. ■ Report black or bloody stools, diarrhea not under control within 24 hours, dry mouth, fever or chills, inability to retain oral fluids due to nausea and vomiting, infections, symptoms of dehydration (e.g., light-headedness, dizziness, fainting), urine changes, or vomiting immediately; each must be treated promptly. Have loperamide (Imodium) available. Dose of loperamide prescribed for late diarrhea is higher than the usual dose recommendation. Limit use at this dose to 48 hours to avoid risk of paralytic ileus. ■ May cause dizziness or visual disturbances (usually within 24 hours of administration); use caution in tasks that require alertness. ■ Compliance with regimen imperative (e.g., taking temperature, obtaining lab work, adequate rest, nourishment, and fluids). ■ Effective birth control required for both women and men; see Maternal/Child. ■ Inform health care professionals of any problems with previous treatments. ■ See Appendix D, p. 1333.
Maternal/child:
Both formulations:
Safety and effectiveness for use in pediatric patients not established.
Conventional irinotecan:
Category D: avoid pregnancy. May cause fetal harm. ■ Discontinue breast-feeding.
Onivyde:
Can cause fetal harm; avoid pregnancy. Advise females of reproductive potential to use effective contraception during treatment and for 1 month after the final dose. Advise males with female partners of reproductive potential to use condoms during treatment and for 4 months after the final dose. ■ Discontinue breast-feeding during treatment and for 1 month after the final dose.
Elderly:
Conventional irinotecan:
Half-life slightly extended. ■ Reduce starting dose by one dose level to 300 mg/M2 in patients 70 years and older in the single-agent, once-every-3-weeks regimen. No change in the starting dose is recommended for elderly patients receiving the weekly dose schedule of irinotecan. See Usual Dose and Dose Adjustments. ■ Risk of early and late diarrhea increased in the elderly. ■ Monitor carefully; may dehydrate more quickly from diarrhea. Begin loperamide therapy promptly. Avoid laxatives.
Onivyde:
No overall differences in safety and effectiveness observed between the elderly and younger patients.
Drug/lab interactions
Interaction of conventional irinotecan and/or ONIVYDE with other drugs has not been adequately studied.
Both formulations:
Concomitant use with CYP3A4 enzyme–inducing anticonvulsants and strong CYP3A4 inducers may increase the metabolism of irinotecan, which decreases concentrations and effectiveness. Avoid use of strong CYP3A4 inducers (e.g., carbamazepine [Tegretol], oxcarbazepine [Trileptal], phenobarbital [Luminal], phenytoin [Dilantin], rifampin [Rifadin], rifabutin [Micobutin], St. John’s wort) if possible. Substitute non–enzyme inducing therapies at least 2 weeks before initiation of irinotecan therapy. ■ CYP3A4 inhibitors (e.g., clarithromycin [Biaxin], indinavir [Crixivan], itraconazole [Sporanox], ketoconazole [Nizoral], lopinavir/ritonavir [Kaletra], nefazodone, nelfinavir [Viracept], ritonavir [Norvir], saquinavir [Invirase], teleprevir [Incivek], voriconazole [VFEND] ) and UGT1A1 inhibitors (e.g., atazanavir [Reyataz], gemfibrozil [Lopid], indinavir [Crixivan], ketoconazole [Nizoral]) may decrease metabolism, which increases serum concentrations and the risk of toxicity. Avoid the use of strong CYP3A4 or UGT1A1 inhibitors if possible. Discontinue at least 1 week before starting irinotecan therapy. ■ Do not administer live virus vaccines to patients receiving antineoplastic drugs.
Conventional irinotecan:
Additive bone marrow suppression may occur with radiation therapy and/or other bone marrow–suppressing agents (e.g., azathioprine, chloramphenicol, melphalan [Alkeran]). Dose reduction may be required. ■ Concurrent administration with irradiation is not recommended. ■ Use caution or withhold diuretics (e.g., furosemide [Lasix]) and laxatives during treatment; may increase risk of dehydration secondary to vomiting and/or diarrhea.
Side effects
Conventional irinotecan:
Myelosuppression (anemia, leukopenia, neutropenia) and diarrhea (“early” [e.g., abdominal cramping or pain, diaphoresis] or “late”) occur in patients and are the most common dose-limiting toxicities with irinotecan administration.
Combination therapy:
Common adverse reactions (greater than 30%) observed in combination therapy are abdominal pain, abnormal bilirubin, alopecia, anemia, anorexia, asthenia, constipation, diarrhea, fever, infection, leukopenia (including lymphocytopenia), mucositis, nausea, neutropenia, pain, thrombocytopenia, and vomiting.
Single-agent therapy:
Common adverse reactions (greater than 30%) observed in single-agent therapy are abdominal pain, alopecia, anemia, anorexia, asthenia, constipation, diarrhea, fever, leukopenia (including lymphocytopenia), nausea, neutropenia, weight loss, and vomiting.
Other reported side effects with irinotecan include abdominal bloating, back pain, chills, confusion, coughing, dehydration, dizziness, dyspepsia, dyspnea, edema, exfoliative dermatitis, flatulence, flushing, headache, hypersensitivity reactions (including anaphylaxis), hyponatremia, hypotension, increased alkaline phosphatase and AST, increased bilirubin, insomnia, interstitial pulmonary disease, muscular contractions or cramps, myocardial infarction, neutropenic fever, neutropenic infection, paresthesia, pneumonia, pulmonary embolism, rash, rhinitis, somnolence, stomatitis, thrombophlebitis, and weight loss may occur. In addition, ileus without preceding colitis has occurred. Renal impairment or failure has occurred, usually in patients who became volume depleted from severe vomiting and/or diarrhea.
Onivyde:
Asthenia, decreased appetite, diarrhea, fatigue, fever, lymphopenia, nausea, neutropenia, stomatitis, and vomiting are most common. The most common serious side effects were acute renal failure, dehydration, diarrhea, fever, nausea, neutropenic fever or neutropenic sepsis, pneumonia, sepsis, septic shock, thrombocytopenia, and vomiting. Diarrhea, vomiting, and sepsis were the most common reasons for discontinuing therapy. Anemia, diarrhea, nausea, and neutropenia were the most common reasons for dose reduction.
Post-marketing:
Asymptomatic elevated pancreatic enzymes (e.g., amylase, lipase), dysarthria (transient), ischemic or ulcerative colitis, megacolon, myocardial ischemic events, pancreatitis.
Antidote
Keep physician informed of all side effects and monitor carefully. Adjust or omit dose as indicated for toxicity; see Dose Adjustments. Treat diarrhea immediately; see Monitor. In the event of an acute onset of new or progressive, unexplained pulmonary symptoms (e.g., cough, dyspnea, fever), interrupt irinotecan and other coprescribed chemotherapeutic agents pending diagnostic evaluation. If interstitial pulmonary disease is diagnosed, discontinue irinotecan and other chemotherapy and initiate appropriate treatment as needed. Administration of whole blood products (e.g., packed RBCs, platelets, leukocytes) and/or blood modifiers (e.g., darbepoetin alfa [Aranesp], epoetin alfa [Epogen], filgrastim [Neupogen, Zarxio], pegfilgrastim [Neulasta], sargramostim [Leukine]) may be indicated to treat bone marrow toxicity. Death may occur from the progression of many side effects. No known antidote for overdose. Maximum supportive care (e.g., to prevent dehydration due to diarrhea and to treat any infectious complications) will help sustain patient in toxicity. If extravasation occurs, flush site with SWFI, elevate the extremity, and apply ice.
Iron dextran injection 
(EYE-ern DEKS-tran in-JEK-shun)
DexFerrum, DexIron  , InFeD, Infufer
, InFeD, Infufer  , Proferdex
, Proferdex
Hematinic
Antianemic
Iron supplement
pH 4.5 to 7
Usual dose
Iron-deficiency anemia:
A test dose is required on the first day. The maximum daily dose is 100 mg/day.
Test dose:
0.5 mL (25 mg) on the first day as a test dose. Wait 1 hour. If no adverse reactions, administer the remainder of the initial therapeutic dose of 1.5 mL (75 mg).
Therapeutic dose:
Repeat the total dose of 2 mL (100 mg)/24 hr daily until results achieved or maximum calculated dosage reached (see dosage charts in literature or formula below). A total calculated dose has been given as an infusion. Though not FDA approved, this method is preferred by some to multiple small-dose infusions or injections. To calculate the total amount of iron dextran (mL) required to restore hemoglobin and to replenish iron stores in adults and pediatric patients weighing over 15 kg (lean body weight [LBW]):
Dose (mL) = 0.0442 (Desired Hb − Observed Hb) × LBW in kg + (0.26 × LBW in kg)
If actual weight is less than LBW or in pediatric patients between 5 and 15 kg, use actual weight in kg. Calculated dose is in mL.
Iron replacement for blood loss:
Dose should represent the equivalent amount of iron represented in blood loss. Begin with a test dose of 0.5 mL (25 mg). Wait 1 hour. If no adverse reactions, calculate the desired dose with the following formula and administer the balance of the replacement dose over 2 to 3 daily doses:
Amount of replacement iron (mg) = Blood loss (mL) × Hematocrit
Calculated dose is in mg; convert to mL before administration.
Formula is based on the approximation that 1 mL of normocytic, normochromic red cells contains 1 mg of elemental iron.
Pediatric dose
Injectable iron not normally used in infants less than 4 months of age. See Maternal/Child.
Iron-deficiency anemia:
Test dose:
0.5 mL (25 mg) for pediatric patients or 0.25 mL (12.5 mg) for infants over at least 5 minutes on the first day. Wait 1 hour. If no adverse reactions, may give remainder of daily dose. Direct IV push is not recommended; diluting with NS for infusion may lower the incidence of phlebitis. If actual weight is less than LBW or for pediatric patients between 5 and 15 kg, use actual weight in kg. The following daily doses have been recommended for IM injection by one source: less than 5 kg, 25 mg; 5 to 10 kg, 50 mg; more than 10 kg, 100 mg. Repeat daily until results achieved or maximum calculated dosage reached (see dosage charts in literature or the formula listed earlier).
Iron replacement for blood loss:
Calculate dose by formula used for adult dose. Dose should represent the equivalent amount of iron represented in blood loss. Begin with a test dose of 0.5 mL (25 mg) for pediatric patients or 0.25 mL (12.5 mg) for infants over at least 5 minutes on the first day. Wait 1 hour. If no adverse reactions, administer the balance of the replacement dose over 2 to 3 daily doses.
Dilution
Given undiluted, or up to the total desired dose may be further diluted in 50 to 1,000 mL NS for infusion. D5W may cause additional local pain and phlebitis.
Filters:
No data available from manufacturer.
Storage:
Store unopened vials at CRT, protect from freezing.
Compatibility
Manufacturer states, “Do not mix with other medications or add to parenteral nutrition solutions.”
Rate of administration
Test dose:
25 mg over 5 minutes (DexFerrum) or over 30 seconds (InFeD). Specific rates of test dose infusions not available for other manufacturers.
IV injection:
If no adverse reactions to the test dose, administer 1 mL (50 mg) or a fraction thereof over 1 minute or more. Extend injection time in pediatric patients.
Infusion:
If no adverse reactions to the test dose, infuse remaining dose over 1 to 8 hours (based on amount of dose, amount of diluent, and patient comfort).
Actions
Iron dextran is removed from the plasma by cells of the reticuloendothelial system, which split the complex into its components of iron and dextran. Iron is immediately bound to protein moieties to form hemosiderin or ferritin, the physiologic forms of iron, or to a lesser extent to transferrin. This iron replenishes hemoglobin and depleted iron stores. Serum ferritin peaks approximately 7 to 9 days after iron dextran administration and slowly returns to baseline after about 3 weeks. Dextran is metabolized or excreted. Negligible amounts of iron are lost via the urinary or alimentary pathways after administration of iron dextran. Some placental transfer of iron dextran may occur. Trace amounts of unmetabolized iron dextran are excreted in breast milk.
Indications and uses
Iron deficiency anemia in patients for whom oral administration is unsatisfactory or impossible; identify and treat the cause of the anemia.
Unlabeled uses:
Iron supplementation for patients taking epoetin alfa (Epogen).
Contraindications
Manifestation of hypersensitivity reactions; any anemia other than iron deficiency.
Precautions
Anaphylactic-type reactions, including fatalities, have been reported. Fatal reactions have occurred both after the test dose and in situations in which the test dose was tolerated. Administer in facilities equipped to monitor the patient and respond to any medical emergency. Patients with a history of drug allergy or multiple drug allergies may be at increased risk for anaphylactic-type reactions. Concomitant use of angiotensin-converting enzyme inhibitors (e.g., enalapril [Vasotec], lisinopril [Zestril]) may also increase the risk of reactions. Facilities for monitoring the patient and responding to any medical emergency must be readily available. ■ Iron dextran products are not clinically interchangeable. They differ in chemical characteristics and may differ in clinical and adverse effects. ■ Large IV doses have been associated with an increased incidence of side effects, including arthralgia, backache, chills, dizziness, fever, headache, malaise, myalgia, nausea, and vomiting. The onset of these side effects is often delayed (1 to 2 days) and symptoms generally subside within 3 to 4 days. Maximum daily recommended dose is 100 mg (2 mL) of undiluted iron dextran. ■ Use with caution in patients with severe liver impairment, cardiovascular disease, or a history of significant allergies and/or asthma. ■ Do not administer during the acute phase of infectious kidney disease. ■ Patients with rheumatoid arthritis may experience increased joint pain and swelling after administration of iron dextran. ■ Administration of parenteral iron therapy should be limited to patients in whom clinical and laboratory investigations have established an iron-deficient state. Unwarranted therapy may cause excess storage of iron and possible exogenous hemosiderosis.
Monitor:
Keep patient lying down after injection to prevent postural hypotension. ■ Observe continuously for a hypersensitivity reaction during an infusion. Monitor vital signs. ■ Monitor hemoglobin, hematocrit, reticulocyte count, total iron-binding capacity (TIBC), and percent of saturation of transferrin as indicated to monitor therapy and iron status. May take up to 3 weeks to see response. ■ Monitor serum ferritin assays in prolonged therapy. Consider possibility of false results for months after injection caused by delayed utilization. ■ In patients undergoing chronic renal dialysis who are receiving iron dextran complex, the correlation of body iron stores and serum ferritin may not be valid.
Patient education:
Promptly report S/S of hypersensitivity (e.g., rash, itching, SOB). ■ Promptly report any other side effects, immediate or delayed.
Maternal/child:
Category C: use only if absolutely necessary in pregnancy, breast-feeding, or childbearing years. ■ Injectable iron not normally used in infants younger than 4 months of age.
Drug/lab interactions
Inhibited by chloramphenicol. ■ Concurrent administration of medicinal iron with dimercaprol (BAL in Oil) will result in the formation of a toxic complex. Either postpone iron therapy or treat severe iron deficiency with transfusions. ■ Effectiveness negated by deferoxamine (Desferal), an iron chelating agent. May be affected by other chelating agents (e.g., edetate disodium). Give iron dextran at least 2 hours after a chelating agent. ■ May cause false serum iron values within 1 to 2 weeks of large doses of iron dextran. ■ May cause a false elevated bilirubin, false decreased calcium, or affect numerous other tests or scans. ■ See Monitor.
Side effects
Backache, dizziness, headache, itching, local phlebitis at injection site, malaise, nausea, rash, shivering, transitory paresthesias.
Major:
Anaphylaxis (fatalities have occurred); arthritic reactivation; dyspnea; febrile episodes; hypotension; leukocytosis; local phlebitis; lymphadenopathy; peripheral vascular flushing, especially with too-rapid injection; tachycardia; urticaria; shock (severe iron toxicity increases vasodilation and venous pooling and decreases circulating blood volume. Results in decreased cardiac output, hypotension, increased peripheral vascular resistance, and shock).
Overdose:
Serum iron levels greater than 300 mcg/dL may indicate iron poisoning. Overdose with iron dextran is unlikely. May result in hemosiderosis, and excess iron may increase susceptibility to infection. If acute toxicity is seen, it may present as:
Early:
Abdominal pain, diarrhea, vomiting.
Late:
Bluish-colored lips, fingernails, and palms of hands; acidosis, drowsiness, shallow and rapid breathing, clammy skin, weak and fast heartbeat, hypotension, hypoglycemia, cardiovascular collapse.
Antidote
Discontinue the drug and notify the physician of early symptoms. For severe symptoms, discontinue drug, treat hypersensitivity reactions, or resuscitate as necessary, and notify physician. Epinephrine (Adrenalin) and diphenhydramine (Benadryl) should always be available. In overdose, monitor CBC, iron studies, vital signs, blood gases, and glucose and electrolytes. Maintain fluid and electrolyte balance. Correct acidosis with sodium bicarbonate. Deferoxamine is an iron chelating agent and may be useful in iron toxicity or overdose. Dialysis will not remove iron alone but will remove the iron deferoxamine complex and is indicated if oliguria or anuria is present.
Iron sucrose
(EYE-ern SOO-kros)
Venofer
Hematinic
Iron supplement
Antianemic
pH 10.5 to 11.1
Usual dose
Dose is expressed in terms of mg of elemental iron. The usual total treatment course of iron sucrose is 1,000 mg. Treatment may be repeated if iron deficiency recurs.
Test dose (optional):
50 mg. Test dose is not required but was administered in some of the clinical trials. May be given at the physician’s discretion.
Adult patients with hemodialysis-dependent chronic kidney disease (HDD-CKD):
100 mg as a slow IV injection or as a 15-minute infusion during consecutive hemodialysis sessions. Administered early during the dialysis session. Repeat as needed to maintain target levels of hemoglobin, hematocrit, and laboratory parameters of iron stores within acceptable limits. Frequency of dosing should be no more than three times weekly.
Adult patients with non–dialysis dependent chronic kidney disease (NDD-CKD):
200 mg as a slow IV injection over 2 to 5 minutes or as an infusion of 200 mg in a maximum of 100 mL of NS over 15 minutes. Administer on 5 different days within a 14-day period to a total cumulative dose of 1,000 mg. Alternately, there is limited experience with administering a 500-mg dose on Day 1 and Day 14 as a 3.5- to 4-hour infusion in a maximum of 250 mL of NS.
Adult patients with peritoneal dialysis–dependent chronic kidney disease (PDD-CKD):
300 mg as an infusion on Day 1 and Day 14. Follow with 400 mg as an infusion on Day 28. A total cumulative dose of 1,000 mg given in 3 doses over 28 days. Each infusion is diluted in a maximum of 250 mL NS and administered over 1.5 to 2.5 hours; see Rate of Administration.
Pediatric dose
Pediatric patients (2 years of age and older) with hemodialysis-dependent chronic kidney disease (HDD-CKD) for iron maintenance treatment:
The dosing for iron replacement treatment in pediatric patients with HDD-CKD has not been established.
Iron maintenance treatment:
0.5 mg/kg. Do not exceed 100 mg/dose. Administer every 2 weeks for 12 weeks; given undiluted by slow IV injection over 5 minutes or diluted in 25 mL of NS and administered over 5 to 60 minutes. Regimen may be repeated if necessary.
Pediatric patients (2 years of age and older) with non–dialysis dependent chronic kidney disease (NDD-CKD) or peritoneal dialysis–dependent chronic kidney disease (PDD-CKD) who are undergoing erythropoietin therapy for iron maintenance treatment:
The dosing for iron replacement treatment in pediatric patients with NDD-CKD or PDD-CKD has not been established.
Iron maintenance treatment:
0.5 mg/kg. Do not exceed 100 mg/dose. Administer every 4 weeks for 12 weeks; given undiluted by slow IV injection over 5 minutes or diluted in 25 mL of NS and administered over 5 to 60 minutes. Regimen may be repeated if necessary.
Dose adjustments
Begin at the low end of the dosing range in elderly patients. Consider the potential for decreased organ function and concomitant disease or drug therapy. ■ Withhold in patients with evidence of tissue iron overload; see Monitor.
Dilution
Available in several sizes of single-dose vials. Check vial carefully and select the size that is closest to the desired dose. All contain 20 mg/mL of elemental iron. May be given undiluted or further diluted in NS. Do not dilute to concentrations below 1 mg/mL.
Test dose (optional):
Adult patients with hemodialysis-dependent chronic kidney disease (HDD-CKD):
100-mg dose may be given undiluted or may be further diluted in a maximum of 100 mL of NS and given as an infusion.
Adult patients with non–dialysis dependent chronic kidney disease (NDD-CKD):
200-mg dose may be given undiluted or may be further diluted in a maximum of 100 mL of NS. The 500-mg dose may be further diluted in a maximum of 250 mL of NS and given as an infusion.
Adult patients with peritoneal dialysis–dependent chronic kidney disease (PDD-CKD):
Each 300- or 400-mg dose must be diluted in a maximum of 250 mL NS and given as an infusion.
Pediatric patients receiving iron maintenance treatment:
A single dose may be given undiluted or diluted in 25 mL of NS and given as an infusion; see Usual Dose.
Filters:
No data available from manufacturer.
Storage:
Store unopened vials in original carton at CRT. Do not freeze. Diluted iron infusions should be used immediately after preparation. Discard any unused portion. When diluted with NS to a concentration of 1 to 2 mg/mL in PVC or non-PVC infusion bags, iron sucrose is stable for 7 days at CRT. When undiluted (20 mg/mL) or diluted with NS to a concentration of 2 to 10 mg/mL and stored in a plastic syringe, iron sucrose is stable for 7 days at CRT or refrigerated.
Compatibility
Manufacturer states, “Do not mix with other medications or add to parenteral nutrition solutions.”
Rate of administration
Too-rapid administration may cause hypotension or symptoms of overdose; see Side Effects.
Test dose (optional):
A single dose over 3 to 10 minutes.
Slow IV injection in adults:
A single undiluted dose over 2 to 5 minutes. In dialysis patients, administer into the dialysis line during the dialysis session.
Infusion in adults:
This method of administration may reduce the risk of hypotensive episodes.
Adult hemodialysis-dependent chronic kidney disease patients (HDD-CKD):
A single 100-mg dose given as a slow IV injection over 2 to 5 minutes. Alternately may be given as an infusion equally distributed over at least 15 minutes.
Adult non–dialysis dependent chronic kidney disease patients (NDD-CKD):
A single 200-mg dose as a slow IV injection over 2 to 5 minutes or as an infusion equally distributed over 15 minutes. Has also been administered as an infusion in a 500-mg dose equally distributed over 3.5 to 4 hours.
Adult peritoneal dialysis–dependent chronic kidney disease patients (PDD-CKD):
A single 300-mg dose equally distributed over 1.5 hours or a single 400-mg dose equally distributed over 2.5 hours.
Pediatric patients receiving iron maintenance treatment:
A single dose as a slow IV injection over 5 minutes or diluted with 25 mL NS and administered as an infusion over 5 to 60 minutes; see Usual Dose.
Actions
An aqueous complex of polynuclear iron (III)-hydroxide in sucrose. Used to replenish the total body iron stores in patients with iron deficiency. Iron is critical for normal hemoglobin synthesis to maintain oxygen transport and necessary for metabolism and synthesis of DNA and various other processes. Following intravenous administration, iron sucrose is dissociated by the reticuloendothelial system into iron and sucrose. Iron distributes into liver, spleen, and bone marrow. Because iron disappearance from serum depends on the need for iron in the iron stores and iron-utilizing tissues of the body, serum clearance of iron is expected to be more rapid in iron-deficient patients as compared to healthy individuals. Half-life of the iron component is 6 hours. The sucrose component is eliminated mainly by urinary excretion. Most iron is stored in the body in hemoglobin, bone marrow, spleen, liver, and ferritin. Small amounts are eliminated in the urine. Significant increases in serum iron and ferritin and significant decreases in total iron-binding capacity occur within 4 weeks of beginning iron sucrose treatment.
Indications and uses
Treatment of iron deficiency anemia in patients with chronic kidney disease (CKD).
Contraindications
Known hypersensitivity to iron sucrose or any of its inactive components. Should not be used in patients with evidence of iron overload or in patients with anemia not caused by iron deficiency.
Precautions
Use only when truly indicated to avoid excess storage of iron. Not recommended for use in patients with iron overload. ■ Potentially fatal hypersensitivity reactions characterized by anaphylactic shock, loss of consciousness, collapse, hypotension, dyspnea, or convulsions have been reported. Medications and equipment for resuscitation must be readily available. ■ Hypotension has been reported frequently and may be related to rate of administration and/or total dose administered. Follow guidelines for dosing and administration. See Usual Dose and Rate of Administration.
Monitor:
Confirm IV placement and avoid extravasation; injection site discoloration has been reported following extravasation. ■ Monitor vital signs during and immediately following administration. Recumbent position during and after administration may help to prevent postural hypotension. Hypotensive effects may be additive to transient hypotension during dialysis and/or from too-rapid rate of administration. ■ Monitor for S/S of hypersensitivity reactions during and after administration for at least 30 minutes and until clinically stable; see Precautions. Reactions may occur after the first dose or subsequent doses of iron sucrose. ■ Periodic monitoring of hematologic and hematinic parameters (hemoglobin, hematocrit, serum ferritin and transferrin saturation) is indicated during parenteral iron replacement therapy. Takes about 4 weeks of treatment to see increased serum iron and ferritin and decreased TIBC (total iron-binding capacity). Transferrin saturation values increase rapidly after IV adminstration of iron sucrose; thus serum iron values may be reliably obtained 48 hours after the last IV dose.
Patient education:
Review any possible reactions to past parenteral iron therapy. ■ Report S/S of a hypersensitivity reaction promptly.
Maternal/child:
Category B: use in pregnancy only if clearly needed. ■ Safety for use during breast-feeding not established. ■ Safety and effectiveness for iron replacement treatment in pediatric patients have not been established. Safety and effectiveness for iron maintenance treatment have been established for pediatric patients 2 years of age and older. ■ Necrotizing enterocolitis in 5 premature infants (weight less than 1,250 Gm) with 2 deaths has been reported in one country in which iron sucrose is approved for use in pediatric patients. No causal relationship to drugs could be established; it may be a complication of prematurity in very-low-birth-weight infants.
Elderly:
Differences in response between elderly and younger patients have not been identified. Lower-end initial doses may be appropriate in the elderly; see Dose Adjustments.
Drug/lab interactions
Drug interactions involving iron sucrose have not been studied. May reduce the absorption of concomitantly administered oral iron preparations; concurrent use not recommended.
Side effects
Adult patients:
Arthralgia, back pain, chest pain, diarrhea, dizziness, headache, hypotension, injection site burning or pain, muscle cramps, nausea, pain in extremity, peripheral edema, pruritus, and vomiting are most common. Other side effects varied according to the type of chronic kidney disease patient who was receiving iron sucrose (e.g., hemodialysis dependent, peritoneal dialysis–dependent, non–dialysis dependent) and included abdominal pain, altered taste, asthenia, catheter site infection, conjunctivitis, cough, dyspnea, ear pain, fecal occult blood positive, feeling abnormal, fever, fluid overload, gout, graft complications, hyperglycemia, hypersensitivity reactions, hypertension, hypoesthesia, hypoglycemia, injection site extravasation, myalgia, nasal congestion, nasopharyngitis, peritoneal infection, pharyngitis, sinusitis, upper respiratory infection.
Pediatric patients:
Arteriovenous fistula thrombosis, cough, dizziness, fever, headache, hypertension, hypotension, nausea, peritonitis, renal transplant, respiratory tract viral infection, and vomiting occurred.
Overdose:
Serum iron levels greater than 300 mcg/dL may indicate iron poisoning. May result in hemosiderosis. Excess iron may increase susceptibility to infection. If acute toxicity is seen, it may present as abdominal and muscle pain, cardiovascular collapse, dizziness, dyspnea, edema, headache, hemosiderosis, hypotension, joint aches, nausea, pale eyes, paresthesia, sedation, vomiting.
Post-marketing:
Life-threatening hypersensitivity reactions (e.g., anaphylactic shock, angioedema, bronchospasm, collapse, convulsions, dyspnea, loss of consciousness, pruritus, rash, shock, wheezing), bradycardia, chromaturia, confusion, CHF, light-headedness, sweating, swelling of joints.
Antidote
Reduce rate of infusion for hypotension or other symptoms. Most symptoms are successfully treated with IV fluids, hydrocortisone sodium succinate (Solu-Cortef), and/or antihistamines (e.g., diphenhydramine [Benadryl]). Volume expanders (e.g., albumin, dextran, hetastarch [Hespan]) may be indicated. Keep physician informed of all side effects. Discontinue drug if severe hypersensitivity reactions occur. Treat hypersensitivity reactions as indicated; may require epinephrine, airway management, oxygen, IV fluids, antihistamines (e.g., diphenhydramine [Benadryl]), corticosteroids (e.g., hydrocortisone sodium succinate [Solu-Cortef]), and pressor amines (e.g., dopamine). Resuscitate as needed. Not removed by dialysis.
Isavuconazonium sulfate
(eye-sah-vew-koh-nah-ZOH-nee-um SUL-fayt)
Cresemba
Antifungal (azole derivative)
Usual dose
Obtain specimens for fungal culture and other relevant lab studies (including histopathology) to isolate and identify causative organism(s) before initiating therapy. Institute antifungal therapy and adjust based on results of culture and lab studies.
Loading dose:
372 mg (1 vial) as an infusion every 8 hours for 6 doses.
Maintenance dose:
372 mg (1 vial) as an infusion once daily. Initiate 12 to 24 hours after the last loading dose. Oral formulation (2 capsules = 372 mg) may be substituted when appropriate.
Dose adjustments
No dose adjustment is required based on age, gender, or race or in patients with mild, moderate, or severe renal impairment, including ESRD. ■ No dose adjustment is required with mild to moderate hepatic impairment.
Dilution
Available as a lyophilized powder containing 372 mg isavuconazonium sulfate (equivalent to 200 mg isavuconazole). Reconstitute 1 vial with 5 mL of SWFI. Gently shake to dissolve the powder completely. Solution should be clear and free of particulates. Withdraw 5 mL from the reconstituted vial and inject into a 250-mL bag of NS or D5W. Final concentration is approximately 1.5 mg/mL. Diluted solution may have visible translucent to white particulates, which will be removed by in-line filtration. Use gentle mixing or roll bag to minimize the formation of particulates. Avoid unnecessary vibration or vigorous shaking of the solution. Do not use a pneumatic transport system.
Filters:
Must be administered using an infusion set that contains a sterile, nonpyrogenic, in-line filter (pore size of 0.2 to 1.2 micrometers).
Storage:
Before use, refrigerate at 2° to 8° C (36° to 46° F). Reconstituted solution should be further diluted and used immediately but may be stored below 25° C for a maximum of 1 hour before further dilution. Diluted solution must be used within 6 hours if kept at RT or may be immediately refrigerated. Complete the infusion within 24 hours. Do not freeze.
Compatibility
Manufacturer states, “Do not infuse isavuconazonium with other intravenous medications.” Compatible only with NS or D5W.
Rate of administration
Do not administer as an IV bolus; for use as a diluted IV infusion only. Administer a single dose as an infusion equally distributed over a minimum of 60 minutes. Flush the IV line before and after each isavuconazonium infusion with NS or D5W. Administer through an infusion set that contains a sterile, nonpyrogenic, in-line filter (pore size of 0.2 to 1.2 micrometers).
Actions
Isavuconazonium is a prodrug of isavuconazole, an azole antifungal drug. Acts by inhibiting the synthesis of ergosterol, a key component of fungal cell membranes. Rapidly hydrolyzed in blood to isavuconazole by esterases, predominantly by butylcholinesterase. Extensively distributed and highly protein bound, predominantly to albumin. Following IV administration, maximum plasma concentrations of the prodrug and inactive products were detectable during infusion and declined rapidly following the end of administration. Mean plasma half-life is 130 hours. CYP3A4, CYP3A5 and, subsequently, uridine diphosphate-glucuronosyltransferase (UGT) are involved in the metabolism of isavuconazole. Primarily excreted as metabolites in urine and feces.
Indications and uses
Treatment of invasive aspergillosis and invasive mucormycosis in patients 18 years of age and older.
Contraindications
Known hypersensitivity to isavuconazole. ■ Coadministration of strong CYP3A4 inhibitors such as ketoconazole (Nizoral) or high-dose ritonavir (Norvir [400 mg every 12 hours]). ■ Coadministration of strong CYP3A4 inducers such as rifampin (Rifadin), carbamazepine (Tegretol), St. John’s wort, or long-acting barbiturates (e.g., phenobarbital [Luminal]). ■ Patients with familial short QT syndrome. ■ See Drug/Lab Interactions.
Precautions
Do not administer as an IV bolus; for use as a diluted IV infusion only. ■ Elevations in liver function tests (e.g., ALT, AST, alkaline phosphatase, total bilirubin) have been reported, were generally reversible, and did not require discontinuation of isavuconazonium. ■ Severe hepatic reactions (e.g., cholestasis, hepatitis, or hepatic failure, including death) have been reported in patients with serious underlying medical conditions (e.g., hematologic malignancy) during treatment with azole antifungal agents, including isavuconazonium. ■ Has not been studied in patients with severe hepatic impairment (Child-Pugh Class C); use in these patients only when benefits outweigh risks. ■ Infusion-related reactions have been reported. ■ Serious hypersensitivity and severe skin reactions, such as anaphylaxis or Stevens-Johnson syndrome, have been reported with other azole antifungal agents. There is no information regarding cross-sensitivity between isavuconazole and other azole antifungal agents. Use caution in patients with hypersensitivity to other azoles. ■ In vitro and animal studies suggest cross-resistance between isavuconazole and other azoles. Relevance of cross-resistance to clinical outcome has not been fully characterized. Patients failing prior azole therapy may require alternative antifungal therapy. ■ See Drug/Lab Interactions.
Monitor:
Obtain specimens for fungal culture and other relevant lab studies (including histopathology) to isolate and identify causative organism(s) before initiating therapy. Institute antifungal therapy and adjust based on results of culture and lab studies. ■ Obtain baseline liver function tests and repeat as necessary. Monitor more frequently in patients who develop abnormal liver function tests and when treating patients with severe hepatic impairment. ■ Monitor for infusion-related reactions (e.g., chills, dizziness, dyspnea, hypoesthesia, hypotension, and paresthesia). ■ Monitor for S/S of hypersensitivity reactions (e.g., hypotension, rash, tightness of the chest, urticaria, wheezing). ■ Monitor for S/S of skin reactions (e.g., rash, blisters).
Patient education:
Review manufacturer’s medication guide. ■ Review of health history and medication profile is imperative. ■ Avoid pregnancy; nonhormonal birth control recommended; see Drug/Lab Interactions. Should pregnancy occur, notify physician and discuss potential hazards. ■ Promptly report S/S of a hypersensitivity/infusion reaction (e.g., itching, hives, shortness of breath, or a serious skin reaction [e.g., rash]).
Maternal/child:
Category C: may cause fetal harm. Use during pregnancy only if the potential benefit to the patient outweighs the risk to the fetus. Report a suspected pregnancy. ■ Discontinue breast-feeding. ■ Safety and effectiveness for use in patients under 18 years of age have not been established.
Elderly:
Response similar to that seen in younger adults.
Drug/lab interactions
Isavuconazole is a sensitive substrate of CYP3A4. CYP3A4 inhibitors or inducers may alter the plasma concentrations of isavuconazole. Isavuconazole is a moderate inhibitor of CYP3A4, a mild inhibitor of P-glycoprotein (P-gp), and an organic cation transporter 2 (OCT2). ■ Coadministration of isavuconazonium with strong CYP3A4 inhibitors such as ketoconazole (Nizoral) or high-dose ritonavir (Norvir [400 mg every 12 hours]) is contraindicated. Strong CYP3A4 inhibitors can significantly increase the plasma concentration of isavuconazole. ■ Coadministration of isavuconazonium with strong CYP3A4 inducers such as rifampin (Rifadin), carbamazepine (Tegretol), St. John’s wort, or long-acting barbiturates (e.g., phenobarbital [Luminal]) is contraindicated. Strong CYP3A4 inducers can significantly decrease the plasma concentration of isavuconazole. ■ Coadministration with lopinavir/ritonavir (Kaletra) can significantly increase the plasma concentration of isavuconazole, and isavuconazole may decrease the antiviral effectiveness of lopinavir/ritonavir. ■ Coadministration with atorvastatin (Lipitor) may increase atorvastatin exposure and toxicity. ■ Coadministration with cyclosporine (Sandimmune), mycophenolate (CellCept), sirolimus (Rapamune), and tacrolimus (Prograf) may increase exposure of these drugs. Monitor drug concentrations of these drugs and/or drug-related toxicities and adjust doses as needed. ■ May increase exposure of midazolam (Versed). Consider dose reduction of midazolam with concomitant administration. ■ May decrease exposure of bupropion (Wellbutrin, Zyban). Dose increase of bupropion may be necessary with coadministration; do not exceed the maximum recommended dose. ■ Concomitant administration with digoxin increases digoxin exposure. Monitor serum digoxin concentrations and adjust dose as necessary.
Side effects
The most commonly reported side effects include back pain; constipation; cough; diarrhea; dyspnea; elevated ALT, AST, alkaline phosphatase, total bilirubin, and GGT; headache; hypokalemia; nausea and vomiting; and peripheral edema. Hepatic adverse drug reactions, infusion-related or hypersensitivity reactions, and embryo-fetal toxicity are considered the most serious. Abdominal pain, acute respiratory failure, anxiety, chest pain, decreased appetite, delirium, dyspepsia, fatigue, hypomagnesemia, hypotension, injection site reaction, insomnia, pruritus, rash, and renal failure have also been reported.
Antidote
Notify physician of all side effects; most will be treated symptomatically. If a hypersensitivity reaction, infusion reaction, or severe cutaneous reaction occurs, discontinue the infusion and treat as indicated. Discontinue isavuconazonium if clinical S/S consistent with liver disease develop that may be attributable to isavuconazonium. No known specific antidote. Not removed by hemodialysis.
Isoproterenol hydrochloride
(eye-so-PROH-ter-ih-nohl hy-droh-KLOR-eyed)
Isuprel
Cardiac stimulant (inotropic/chronotropic)
Bronchodilator
Antiarrhythmic
pH 3.5 to 4.5
Usual dose
In all situations, adjust the rate of infusion based on HR, CVP, BP, respiratory rate, and urine output.
| Recommended Isoproterenol Dose for Adults with Atropine-Resistant Hemodynamically Significant Bradycardia, Heart Block, Adams-Stokes Attacks, and Cardiac Arrest | |||
| Route of Administration | Preparation of Dilution | Initial Dose | Subsequent Dose Range* |
| Bolus intravenous injection | Dilute 1 mL (0.2 mg) to 10 mL with NS or D5W | 0.02 to 0.06 mg (1 to 3 mL of diluted solution) | 0.01 to 0.2 mg (0.5 to 10 mL of diluted solution) |
| Intravenous infusion | Dilute 10 mL (2 mg) in 500 mL D5W (4 mcg/mL) | 5 mcg/min (1.25 mL of diluted solution per minute) | |
| Intracardiac | Use solution 0.2 mg/mL undiluted | 0.02 mg (0.1 mL) | |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


