(dah-KAR-bah-zeen)
DTIC, DTIC-Dome
Antineoplastic (alkylating agent)
pH 3 to 4
Usual dose
Malignant melanoma:
2 to 4.5 mg/kg of body weight/24 hr for 10 days. May be repeated at 4-week intervals. May administer 250 mg/M2 daily for 5 days. Repeat in 3 weeks. Has proved as effective in lesser doses as in larger doses. Individualized response determines dosage of subsequent treatments.
Hodgkin’s disease:
150 mg/M2/24 hr for 5 days. Repeat every 4 weeks. Used in combination with other drugs in a specific regimen. An alternate regimen is 375 mg/M2 on Days 1 and 15 every 4 weeks or 100 mg/M2/day for 5 days. Given as part of a specific protocol.
Dose adjustments
Dose (mg/kg) based on average weight in presence of edema or ascites. ■ Used with other antineoplastic drugs and radiation therapy in reduced doses to achieve tumor remission. ■ Dose reduction may be required in impaired liver and renal function.
Dilution
Specific techniques required; see precautions.
Each 100-mg vial is diluted with 9.9 mL (200 mg with 19.7 mL) of SWFI (10 mg/mL). Further dilution in 50 to 250 mL of D5W or NS for infusion is preferred. May be given through Y-tube or three-way stopcock of infusion set through a free-flowing IV.
Storage:
Discard in 6 to 8 hours if kept at room temperature. Reconstituted solution stable for 72 hours, diluted solution for 24 hours if refrigerated at 4° C (39° F).
Compatibility (underline indicates conflicting compatibility information)
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
One source suggests the following compatibilities:
Additive:
Ondansetron (Zofran).
Y-site:
Amifostine (Ethyol), aztreonam (Azactam), doxorubicin liposomal (Doxil), etoposide phosphate (Etopophos), filgrastim (Neupogen), fludarabine (Fludara), granisetron (Kytril), heparin, melphalan (Alkeran), ondansetron (Zofran), paclitaxel (Taxol), palonosetron (Aloxi), sargramostim (Leukine), teniposide (Vumon), thiotepa, vinorelbine (Navelbine).
Rate of administration
Total dose over 30 to 60 minutes. More rapid rate may cause severe venous irritation.
Actions
An antineoplastic agent. Exact mechanism of action is not known; may inhibit DNA and RNA synthesis. It is an alkylating agent, cell cycle phase nonspecific. Probably localizes in the liver and is excreted in the urine.
Indications and uses
Metastatic malignant melanoma. ■ Hodgkin’s disease. ■ Soft-tissue sarcomas.
Unlabeled uses:
Treatment of malignant pheochromocytoma with cyclophosphamide and vincristine. ■ Treatment of metastatic malignant melanoma with tamoxifen.
Contraindications
Known hypersensitivity to dacarbazine.
Precautions
Follow guidelines for handling cytotoxic agents. See Appendix A, p. 1331. ■ Administered by or under the direction of the physician specialist. ■ Bone marrow suppression is the most common toxicity. ■ Hepatic necrosis has been reported. ■ Consider potential for therapeutic benefit versus risk for toxicity. ■ Use caution in impaired liver and renal function.
Monitor:
Determine absolute patency of vein; a stinging or burning sensation indicates extravasation; severe cellulitis and tissue necrosis will result. Discontinue injection; use another vein. ■ Monitor bone marrow function, white and RBC count, and platelet count frequently. ■ Nausea and vomiting may be reduced by restricting oral intake of fluid and foods for 4 to 6 hours before administration. Use prophylactic antiemetics. ■ Be alert for signs of bone marrow suppression, bleeding, or infection. ■ Monitor for thrombocytopenia (platelet count less than 50,000/mm3). Initiate precautions to prevent excessive bleeding (e.g., inspect IV sites, skin, and mucous membranes; use extreme care during invasive procedures; test urine, emesis, stool, and secretions for occult blood). ■ Prophylactic antibiotics may be indicated pending results of C/S in a febrile neutropenic patient.
Patient education:
Protect skin surfaces; may cause photosensitive skin reactions. ■ Nonhormonal birth control recommended. ■ Report burning or stinging at IV site promptly. ■ See Appendix D, p. 1333.
Maternal/child:
Category C: safety for use in pregnancy or breast-feeding and in men and women capable of conception not established. ■ Carcinogenic and teratogenic in animals. ■ Discontinue breast-feeding.
Elderly:
Consider age-related organ impairment; toxicity may be increased.
Drug/lab interactions
Do not administer any live virus vaccines to patients receiving antineoplastic drugs. ■ Inhibited by phenobarbital and phenytoin (Dilantin). ■ Potentiates allopurinol. ■ Effects of dacarbazine may be increased with ciprofloxacin (Cipro), isoniazid (INH), fluvoxamine (Luvox), ketoconazole (Nizoral), miconazole (Monistat), and norfloxacin (Noroxin). Effects may be decreased with carbamazepine (Tegretol), phenobarbital, and rifampin (Rifadin).
Side effects
Leukopenia and thrombocytopenia may be serious enough to cause death. Alopecia, anaphylaxis, anorexia, facial flushing, facial paresthesias, fever, hepatotoxicity, malaise, myalgia, nausea, skin necrosis, vomiting.
Antidote
Notify physician of all side effects. Most will be treated symptomatically. Bone marrow suppression may require temporary or permanent withholding of treatment. Administration of whole blood products (e.g., packed RBCs, platelets, leukocytes) and/or blood modifiers (e.g., darbepoetin alfa [Aranesp], epoetin alfa [Epogen], filgrastim [Neupogen, Zarxio], pegfilgrastim [Neulasta], sargramostim [Leukine]) may be indicated to treat bone marrow toxicity. There is no specific antidote. Supportive therapy as indicated will help sustain the patient in toxicity. For extravasation, elevate extremity; consider injection of long-acting dexamethasone (Decadron LA) throughout extravasated tissue. Use a 27- or 25-gauge needle. Apply moist, warm compresses.
Dactinomycin 
(dack-tin-oh-MY-sin)
Cosmegen
Antineoplastic (antibiotic)
pH 5.5 to 7
Usual dose
Dose will depend on tolerance of the patient, the size and location of the tumor, and the use of other forms of therapy. Calculate each dose carefully before administration. Calculation of the dose for obese or edematous patients should be based on body surface area. The dose intensity per 2-week cycle for adult and pediatric patients should not exceed 15 mcg/kg/day (0.015 mg/kg/day) or 400 to 600 mcg/M2/day (0.4 to 0.6 mg/M2/day) for 5 days.
Wilms’ tumor, childhood rhabdomyosarcoma, and Ewing’s sarcoma:
15 mcg/kg/day for 5 days, not to exceed 0.5 mg/day (500 mcg/day). May be administered in various combinations and schedules with other chemotherapeutic agents.
Metastatic nonseminomatous testicular cancer:
1,000 mcg/M2 (1 mg/M2) on Day 1 as part of a combination regimen with cyclophosphamide, bleomycin, vinblastine, and cisplatin.
Gestational trophoblastic neoplasia:
12 mcg/kg/day for 5 days as a single agent or 500 mcg on Days 1 and 2 as part of a combination regimen with etoposide, methotrexate, folinic acid, vincristine, cyclophosphamide, and cisplatin.
Pediatric dose
Same as Usual Dose for adults; see Contraindications.
Dose adjustments
Calculate dose based on body surface area in presence of edema or ascites. ■ Used with other antineoplastic drugs in reduced doses to achieve tumor remission. ■ Reduce dose of dactinomycin and radiation therapy when used concurrently, if either has been used previously, or if previous chemotherapy has been employed. ■ Dose selection should be cautious in the elderly. Consider potential for decreased cardiac, hepatic, and renal function, and concomitant disease or drug therapy; see Elderly.
Dilution
Specific techniques required; see Precautions. Highly toxic. Both powder and solution must be handled with care. Dilute each 0.5-mg vial with 1.1 mL of preservative-free SWFI (0.5 mg/mL). Reconstituted product is a clear, gold-colored solution. SWFI with preservative (benzyl alcohol or paraben) will cause precipitation. Very corrosive to soft tissue. Use sterile two-needle technique if given by IV injection; one needle to dilute and withdraw and one needle to inject into the vein (rinse with blood or IV solution before removing). May be given by IV injection, through the Y-tube or three-way stopcock of a free-flowing infusion of D5W or NS, or further diluted in one of the above solutions for infusion to a final concentration of 10 mcg/mL or higher.
Filters:
Manufacturer states, “Use of some in-line cellulose ester membrane filters have resulted in loss of potency.” Another source suggests no loss of drug potency with a 5-micron stainless steel depth filter.
Storage:
Store at 25° C (77° F). Protect from light and humidity. Prepared product must be used within 4 hours of initial reconstitution when stored at ambient RT. Discard any unused portion.
Compatibility
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
Forms a precipitate with SWFI that contains preservatives. Cellulose ester membrane filters may reduce dose by partial removal of dactinomycin.
One source suggests the following compatibilities:
Y-site:
Allopurinol (Aloprim), amifostine (Ethyol), aztreonam (Azactam), etoposide phosphate (Etopophos), fludarabine (Fludara), gemcitabine (Gemzar), granisetron (Kytril), melphalan (Alkeran), ondansetron (Zofran), sargramostim (Leukine), teniposide (Vumon), thiotepa, vinorelbine (Navelbine).
Rate of administration
IV injection:
A single dose over 2 to 3 minutes.
IV infusion:
A single dose over 10 to 15 minutes.
Actions
A highly toxic antibiotic antineoplastic agent, cell cycle phase nonspecific. Cytotoxic, it interferes with cell division by binding DNA to slow production of RNA. Found in high concentrations in the kidney, liver, and spleen. Does not penetrate the blood-brain barrier. Minimally metabolized. Elimination half-life is approximately 36 hours. Excreted as unchanged drug in bile and urine.
Indications and uses
As part of a combination chemotherapy and/or multimodality regimen for treatment of Wilms’ tumor, childhood rhabdomyosarcoma, Ewing’s sarcoma, and metastatic nonseminomatous testicular cancer. ■ Alone or in combination with other chemotherapeutic agents for the treatment of gestational trophoblastic neoplasia.
Unlabeled uses:
Treatment of osteosarcoma, malignant melanoma, Paget’s disease of the bone.
Contraindications
Exposure to chickenpox, herpes zoster, known sensitivity to dactinomycin, infants under 6 to 12 months of age.
Precautions
Follow guidelines for handling cytotoxic agents. Review guidelines before handling, and follow diligently. See Appendix A, p. 1331. ■ For IV use only. Do not administer IM or SC. ■ Highly toxic; both the powder and solution must be handled and administered with care. Inhalation of dust or vapors and contact with skin or mucous membranes, especially those of the eyes, must be avoided. If eye contact occurs, rinse for at least 15 minutes with water, saline or a balanced salt ophthalmic solution and then seek immediate ophthalmologic consultation. If skin contact occurs, remove contaminated clothing and rinse area for 15 minutes. Medical attention should be sought immediately and clothes should be destroyed. ■ Administered by or under the direction of a physician specialist. ■ In general, dactinomycin should not be administered concomitantly with radiation therapy in the treatment of Wilms’ tumor unless the benefit outweighs the risk. Hepatomegaly and elevated AST levels have been reported. ■ Hepatic veno-occlusive disease that may be associated with intravascular clotting disorder and multiorgan failure has been reported. Pediatric patients younger than 48 months of age may be at increased risk. ■ May have increased incidence of second primary tumors following treatment with dactinomycin and radiation. Long-term follow-up of cancer survivors is indicated.
Monitor:
Very corrosive to soft tissue; determine absolute patency of vein. If extravasation occurs, severe damage to soft tissue will result. A stinging or burning sensation indicates extravasation; severe cellulitis and tissue necrosis will result. Discontinue injection; use another vein. Close observation and reconstructive surgery consultation are recommended. ■ Monitor renal, hepatic, and bone marrow function frequently. ■ Except for immediate nausea and vomiting, side effects may not appear for 2 to 4 days and may not peak for 1 to 2 weeks. Always observe closely. Use prophylactic antiemetics. ■ If stomatitis, diarrhea, or severe hematopoietic depression appears, discontinue therapy until the patient has recovered. ■ An increased incidence of GI toxicity, bone marrow suppression, and skin and mucosal reactions has been reported when dactinomycin is administered in combination with radiation therapy. ■ Monitor for thrombocytopenia (platelet count less than 50,000/mm3). Initiate precautions to prevent excessive bleeding (e.g., inspect IV sites, skin, and mucous membranes; use extreme care during invasive procedures; test urine, emesis, stool, and secretions for occult blood). ■ Allopurinol, increased fluid intake, and alkalinization of urine may be required to reduce uric acid levels. ■ Observe closely for signs of infection. Prophylactic antibiotics may be indicated pending results of C/S in a febrile neutropenic patient.
Patient education:
Nonhormonal birth control recommended. ■ Report burning or stinging at IV site promptly. ■ See Appendix D, p. 1333.
Maternal/child:
Category D: avoid pregnancy. May produce teratogenic effects on the fetus; use caution in men and women capable of conception. ■ Discontinue breast-feeding. ■ Greater frequency of toxic effects seen in infants. See Contraindications and Precautions.
Elderly:
Response similar to that in younger adults; however, recent studies suggest elderly may be at increased risk for myelosuppression; see Dose Adjustments.
Drug/lab interactions
Dactinomycin potentiates the effects of radiation therapy; use with caution in patients receiving radiation therapy. Reduced doses are indicated with simultaneous use; risk of GI toxicity and myelosuppression increased. ■ Dactinomycin alone may reactivate erythema from previous radiation therapy. ■ Do not administer live virus vaccines to patients receiving antineoplastic drugs. ■ Inhibits action of penicillin. ■ See Dose Adjustments. ■ May interfere with bioassay procedures used in determining antibacterial drug levels.
Side effects
Toxic reactions are frequent, may be severe, and may be dose limiting; however, the severity of toxicity varies markedly and is only partly dependent on the dose administered. Abdominal pain, acne, alopecia, anaphylaxis, anorexia, bone marrow suppression (e.g., anemia, aplastic anemia, agranulocytosis, febrile neutropenia, leukopenia, neutropenia, thrombocytopenia), cheilitis, diarrhea, dysphagia, erythema flare-up, erythema multiforme, esophagitis, fatigue, fever, GI ulceration, hypocalcemia, lethargy, liver toxicity (ascites, hepatitis, hepatic failure with reports of death, hepatic veno-occlusive disease, hepatomegaly, and liver function test abnormalities), malaise, myalgia, nausea, pharyngitis, pneumonitis, proctitis, sepsis (including neutropenic sepsis), skin eruptions, ulcerative stomatitis, vomiting.
Post-marketing:
Stevens-Johnson syndrome, toxic epidermal necrolysis.
Antidote
Any side effect can result in death. Notify the physician of all side effects. Most will be treated symptomatically. Bone marrow suppression may require withholding dactinomycin until recovery occurs. No specific antidote. Supportive therapy as indicated will help sustain the patient in toxicity. Administration of whole blood products (e.g., packed RBCs, platelets, leukocytes) and/or blood modifiers (e.g., darbepoetin alfa [Aranesp], epoetin alfa [Epogen], filgrastim [Neupogen, Zarxio], pegfilgrastim [Neulasta], sargramostim [Leukine]) may be indicated to treat bone marrow toxicity. For extravasation, discontinue immediately and elevate extremity. Apply ice to site four times daily for 3 days. Close observation and reconstructive surgery consultation recommended.
Dalbavancin
(DAL-ba-VAN-sin)
Dalvance
Antibacterial (lipoglycopeptide)
Usual dose
1,500 mg. May be administered as a single dose or as a two-dose regimen with an initial dose of 1,000 mg followed 1 week later by 500 mg. Administer as an infusion over 30 minutes.
Dose adjustments
Dosage should be adjusted in patients with renal impairment as outlined in the following chart.
| Dosage of Dalbavancin in Patients with Renal Impairment | ||
| Estimated CrCl* | Dalbavancin Single-Dose Regimen† | Dalbavancin Two-Dose Regimen† |
| ≥30 mL/min or on regular hemodialysis | 1,500 mg | 1,000 mg followed 1 week later by 500 mg |
| <30 mL/min and not on regular hemodialysis | 1,125 mg | 750 mg followed 1 week later by 375 mg |

*As calculated using the Cockcroft-Gault formula.
■ No dose adjustment is recommended for patients receiving regularly scheduled hemodialysis, and dalbavancin can be administered without regard to the timing of hemodialysis. ■ No dose adjustment is recommended for patients with mild hepatic impairment (Child-Pugh Class A). ■ Use caution when prescribed for patients with moderate or severe hepatic impairment (Child-Pugh Class B or C). No data are available to determine appropriate dosing in these patients. ■ No dose adjustment based on age or gender; see Elderly.
Dilution
Available in single-use vials containing 500 mg of dalbavancin as a lyophilized powder. Reconstitute each 500-mg vial with 25 mL of SWFI or D5W. To avoid foaming, alternate between gentle swirling and inversion of the vial until contents are completely dissolved. Do not shake. Concentration is 20 mg/mL. Solution should be clear and colorless to yellow. Do not use if particulate matter remains. Transfer the required dose to an IV bag or bottle containing D5W. Diluted solution must have a final concentration of 1 mg/mL to 5 mg/mL.
Filters:
Specific information not available.
Storage:
Vials may be stored at CRT. Reconstituted vials and/or fully diluted solutions may be stored at RT or refrigerated at 2° to 8° C (36° to 46° F). Do not freeze. Total time from reconstitution to dilution to administration should not exceed 48 hours.
Compatibility
Manufacturer states, “Do not co-infuse dalbavancin with other medications or electrolytes. Saline-based infusion solutions may cause precipitation and should not be used. The compatibility of reconstituted dalbavancin with IV medications, additives, or substances other than D5W has not been established.”
Rate of administration
A single dose as an infusion equally distributed over 30 minutes. If a common IV line is used to administer other drugs in addition to dalbavancin, flush the IV line before and after each dalbavancin infusion with D5W. Too-rapid infusion may cause “red-man syndrome,” including flushing of the upper body, pruritus, rash, and/or urticaria. Temporarily stop or slow the infusion as indicated.
Actions
Dalbavancin is a semi-synthetic lipoglycopeptide antibacterial drug. It interferes with cell wall synthesis by binding to the d-alanyl-d-alanine terminus of the stem pentapeptide in nascent cell wall peptidoglycan, thus preventing cross-linking. In vitro, dalbavancin is bactericidal against Staphylococcus aureus and Streptococcus pyogenes at concentrations similar to those sustained throughout treatment; see Indications. 93% bound to plasma proteins, primarily to albumin. Mean concentrations achieved in skin blister fluid remain above 30 mg/L up to 7 days after dosing. Effective half-life is approximately 346 hours. Excreted in feces and urine as unchanged drug and as a metabolite.
Indications and uses
Treatment of adult patients with acute bacterial skin and skin structure infections (ABSSSI) caused by susceptible isolates of designated gram-positive microorganisms, including Staphylococcus aureus (both methicillin-susceptible [MSSA] and methicillin-resistant [MRSA] strains).
Contraindications
Known hypersensitivity to dalbavancin. No data available on cross-reactivity between dalbavancin and other glycopeptides, including vancomycin.
Precautions
Serious hypersensitivity (anaphylactic) and skin reactions have been reported. Check history of previous hypersensitivity reactions to glycopeptides (e.g., oritavancin [Orbactiv], telavancin [Vibativ], vancomycin). Exercise caution in patients with a history of glycopeptide allergy; cross-sensitivity is possible. ■ Infusion-related reactions have been reported; see Rate of Administration. ■ Specific sensitivity studies are indicated to determine susceptibility of the causative organism to dalbavancin. ■ To reduce the development of drug-resistant bacteria and maintain its effectiveness, dalbavancin should be used to treat only those infections proven or strongly suspected to be caused by bacteria. ■ Clostridium difficile–associated diarrhea (CDAD) has been reported for nearly all systemic antibacterial agents and may range in severity from mild diarrhea to fatal colitis. Consider in patients who present with diarrhea during or after treatment with dalbavancin. ■ ALT elevations greater than three times the ULN occurred in some patients with normal baseline transaminase levels before treatment.
Monitor:
Obtain baseline SCr. ■ Monitor for S/S of hypersensitivity (e.g., hypotension, rash, urticaria, tightness of the chest, wheezing). ■ Monitor for S/S of an infusion reaction; see Rate of Administration. ■ See Precautions; baseline liver function studies may be indicated.
Patient education:
Promptly report S/S of a hypersensitivity reaction (e.g., hives, rash, shortness of breath, wheezing) or an infusion reaction (e.g., flushing of the upper body, pruritus, rash, and/or urticaria). ■ Promptly report diarrhea or bloody stools that occur during treatment or up to several months after an antibiotic has been discontinued; may indicate CDAD and require treatment.
Maternal/child:
Use during pregnancy only if the potential benefit outweighs the possible risk to the fetus. ■ Use caution during breast-feeding. ■ Safety and effectiveness for use in pediatric patients not established.
Elderly:
Consider age-related renal impairment, monitoring of renal function, and dose with caution; see Dose Adjustments.
Drug/lab interactions
Clinical drug-drug interaction studies have not been conducted. ■ There is minimal potential for drug-drug interactions between dalbavancin and substrates, inhibitors, and inducers of cytochrome P450 enzymes. ■ Dalbavancin pharmacokinetics were not affected by coadministration of acetaminophen (Ofirmev), aztreonam (Azactam), fentanyl, furosemide (Lasix), metronidazole (Flagyl), midazolam (Versed), proton pump inhibitors (e.g., esomeprazole [Nexium], lansoprazole [Prevacid], omeprazole [Prilosec], pantoprazole [Protonix]), and simvastatin (Zocor). ■ In vitro, dalbavancin demonstrated synergistic interactions with oxacillin and did not demonstrate antagonistic or synergistic interactions with aztreonam (Azactam), clindamycin (Cleocin), daptomycin (Cubicin), gentamicin, levofloxacin (Levaquin), linezolid (Zyvox), quinupristin/dalfopristin (Synercid), rifampin (Rifadin), or vancomycin. Clinical significance unknown.
Side effects
Most common side effects reported are diarrhea, headache, and nausea. Hypersensitivity and/or infusion reactions may be severe. Increased ALT levels (reversible), pruritus, rash, and vomiting were also reported. Many other side effects occurred in fewer than 2% of patients.
Antidote
Notify physician of any side effects. Discontinue the drug if indicated. Treat hypersensitivity reactions as indicated (e.g., diphenhydramine [Benadryl], epinephrine [Adrenalin], albuterol) and resuscitate as necessary. Temporarily discontinue or slow infusion for infusion-related reactions. Mild cases of CDAD may respond to discontinuation of dalbavancin. Treat CDAD with fluids, electrolytes, protein supplements, and oral vancomycin (Vancocin) or metronidazole (Flagyl) as indicated. In severe cases, surgical evaluation may be indicated. Less than 6% of the recommended dose of dalbavancin is removed by hemodialysis.
Dantrolene sodium
(DAN-troh-leen SO-dee-um)
Dantrium, Revonto ■ Ryanodex
Skeletal muscle relaxant (direct acting)
pH 9.5 ■ 10.3
Usual dose
In patients known to be susceptible to malignant hyperthermia (MH), oral dantrolene may be used prophylactically preoperatively. Oral or IV therapy should be used postoperatively for 1 to 3 days following IV treatment for MH crisis. Postoperative dosing is indicated after emergency treatment to prevent recurrence of the manifestations of MH.
Prophylactic dose: Dantrium, revonto:
2.5 mg/kg as an infusion. Begin administration 11/4 hours before anesthesia, and administer over 1 hour. Oral dantrolene may be used.
Ryanodex:
2.5 mg/kg as an IV injection over at least 1 minute. Begin administration 11/4 hours before surgery. If surgery is prolonged, administer additional individualized doses during anesthesia and surgery as needed.
All formulations:
Avoid agents that trigger MH (e.g., general anesthetics and depolarizing neuromuscular blocking agents [succinylcholine]).
Therapeutic or emergency dose: All formulations:
Discontinue all anesthetic agents at the first sign of a malignant hyperthermia reaction. Administration of 100% oxygen is recommended.
1 mg/kg of body weight as an initial dose as a rapid IV push. Repeat as necessary until symptoms subside or a cumulative dose of 10 mg/kg is reached. Entire regimen may be repeated if symptoms reappear. Dose required depends on degree of susceptibility to malignant hyperthermia, length of time of exposure to triggering agent, and time lapse between onset of crisis and beginning of treatment.
Post-crisis follow-up:
An oral dose of 4 to 8 mg/kg/day for 1 to 3 days to prevent recurrences. If oral dosing not feasible, begin IV dose at 1 mg/kg and individualize by increasing based on patient response.
Pediatric dose
Prophylactic, therapeutic, and post-crisis follow-up doses are the same as for adults; see Maternal/Child.
Dose adjustments
Dose selection should be cautious in the elderly. Reduced doses may be indicated based on the potential for decreased organ function and concomitant disease or drug therapy.
Dilution
Dantrium, revonto:
Each 20 mg must be diluted with 60 mL SWFI without a bacteriostatic agent. Shake until solution is clear. May be administered through a Y-tube or three-way stopcock of infusion tubing. If large volumes will be used, transfer to plastic infusion bags; do not use glass bottles; see Compatibility.
Ryanodex:
Reconstitute each 250-mg vial with 5 mL of SWFI without a bacteriostatic agent. Shake the vial to ensure an orange-colored uniform suspension. Do not reconstitute with D5W or NS; see Compatibility. May be administered directly into an indwelling catheter or through a Y-tube or three-way stopcock of a free-flowing infusion of D5W or NS.
Storage: All formulations:
Store undiluted vials at CRT and protect from light. Store diluted solution at CRT and protect from direct light. Discard after 6 hours.
Compatibility
Dantrium, revonto:
Manufacturer states, “D5W, NS, and acidic solutions are not compatible and should not be used.” May form a precipitate with glass bottles; use of plastic IV bags recommended.
Ryanodex:
Do not dilute or transfer the reconstituted suspension to another container to infuse the product.
Rate of administration
Prophylactic dose: Dantrium, revonto:
A single dose as an infusion distributed over 1 hour.
Ryanodex:
A single dose as an IV injection over at least 1 minute. If administering into an indwelling catheter without a free-flowing IV, flush line after administration of Ryanodex to ensure there is no residual drug left in the catheter.
Therapeutic or emergency dose: All formulations:
Each single dose should be given by rapid continuous IV push. Follow immediately with subsequent doses as indicated.
Actions
A direct-acting skeletal muscle relaxant. Inhibits excitation-contraction coupling by interfering with the release of the calcium ion from the sarcoplasmic reticulum to reverse the physiologic cause of malignant hyperthermia and produce relaxation. The addition of dantrolene to the “triggered” malignant hyperthermic muscle cell may re-establish a normal level of ionized calcium in the myoplasm. Physiologic, metabolic, and biochemical changes associated with the malignant hyperthermia crisis may be reversed or attenuated. Has no appreciable effect on cardiovascular or respiratory function. Onset of action is prompt. Half-life of Dantrium and Revonto is 4 to 8 hours. Half-life of Ryanodex is 8.5 to 11.4 hours. Metabolized in the liver and excreted in urine. Readily crosses the placental barrier. Secreted in breast milk.
Indications and uses
Treatment of malignant hyperthermia in conjunction with appropriate supportive measures. ■ Prevention of malignant hyperthermia in patients at high risk.
Contraindications
None.
Precautions
Use caution in patients with impaired pulmonary or cardiac function or history of liver disease. ■ Discontinue all anesthetic agents immediately when onset of malignant hyperthermia is recognized. Administration of 100% oxygen is recommended. ■ Hepatotoxicity has been reported with oral dantrolene. ■ Not indicated for use in patients with neuroleptic malignant syndrome (NMS).
Monitor:
S/S of malignant hyperthermia crises include central venous desaturation, cyanosis and mottling of the skin, hypercarbia, increased utilization of anesthesia circuit carbon dioxide absorber, metabolic acidosis, skeletal muscle rigidity, tachycardia, tachypnea and, in many cases, fever. ■ Monitor ECG, vital signs, electrolytes, and urine output continuously. ■ Oxygen needs are increased. ■ Manage metabolic acidosis. ■ Institute cooling measures. ■ Diuretics may be required to prevent or treat late kidney injury due to myoglobinuria. Consider amount of mannitol present in Dantrium or Revonto formulations. (Ryanodex does not contain a sufficient amount of mannitol to maintain diuresis.) ■ Confirm absolute patency of vein; avoid extravasation. High pH may cause tissue necrosis. ■ Associated with skeletal muscle weakness; see Patient Education. ■ Ensure adequate ventilation; has been associated with dyspnea, respiratory muscle weakness, and decreased inspiratory capacity. ■ Assess patients for difficulty swallowing and choking. ■ Monitor hepatic function, including ALT, AST. ■ Somnolence and dizziness may persist for up to 48 hours postdose. Ambulate with assistance.
Patient education:
May experience decreased grip strength, weakness in leg muscles, and light-headedness postoperatively. May persist for 48 hours. ■ Request assistance for ambulation. ■ Use caution when eating; choking and difficulty swallowing have been reported on day of administration. ■ Avoid alcohol and other CNS depressants (e.g., diazepam [Valium]). ■ Avoid tasks that require alertness. ■ Promptly report bloody or tarry stools, itching, jaundice (yellow color) of eyes and skin, or skin rash.
Maternal/child:
Category C: embryocidal in animal studies. Use during pregnancy only if potential benefit justifies potential risk to the fetus. ■ Discontinue breast-feeding. ■ Safety and effectiveness for use in pediatric patients have been established. Dose is the same as for adults.
Elderly:
Differences in responses between the elderly and younger patients have not been identified. Dose selection should be cautious; see Dose Adjustments.
Drug/lab interactions
All formulations:
Avoid concurrent use of calcium channel blockers (e.g., diltiazem [Cardizem]) and dantrolene. Cardiovascular collapse, arrhythmias, and hyperkalemia have been reported. ■ Ability to bind to plasma proteins inhibited by warfarin (Coumadin) and clofibrate (Atromid-S); increased by tolbutamide (Orinase). ■ Phenobarbital (Luminal) and diazepam (Valium) do not affect dantrolene sodium metabolism.
Dantrium and revonto:
May potentiate vecuronium-induced neuromuscular blockade. ■ Binding to plasma protein is not significantly altered by diazepam [Valium], diphenylhydantoin, or phenylbutazone (Butazolidin).
Ryanodex:
May potentiate the neuromuscular block when given with muscle relaxants. ■ May potentiate the effects of antipsychotic agents (e.g., pimozide [Orap], clozapine [Clozaril]) and antianxiety agents (e.g., diazepam [Valium]) on the central nervous system. ■ Concomitant use of sedative agents may increase the risk of somnolence and dizziness.
Side effects
Dizziness, drowsiness, loss of grip strength, and weakness in the legs are most common. Other reported side effects include erythema, hypersensitivity reactions (including anaphylaxis), injection site reactions, nausea, pulmonary edema, thrombophlebitis, tissue necrosis secondary to extravasation, urticaria.
Antidote
No specific antidote is available or needed when used correctly. Notify physician and initiate supportive measures (ensure adequate airway and ventilation, monitor ECG) in overdosage. Large amounts of IV fluids may be needed to prevent crystalluria. Treat anaphylaxis and resuscitate as necessary. Value of dialysis in overdose is not known.
Daptomycin
(dap-toe-MY-sin)
Cubicin
Antibacterial (cyclic lipopeptide)
Usual dose
Complicated skin and skin structure infections:
4 mg/kg once every 24 hours for 7 to 14 days. Do not administer more frequently than once daily.
Staphylococcus aureus bloodstream infections (bacteremia), including those with right-sided endocarditis:
6 mg/kg once every 24 hours for 2 to 6 weeks. Duration of treatment is dependent on diagnosis. Safety data for use more than 28 days is limited. Do not administer more frequently than once daily.
Dose adjustments
Dose adjustment required in patients with severe renal impairment. In patients with CrCl less than 30 mL/min, including patients undergoing hemodialysis or CAPD, administer a single dose (4 or 6 mg/kg) every 48 hours. If possible, administer dose following completion of hemodialysis on hemodialysis days. ■ No specific dose adjustments required based on age, gender, obesity, or mild to moderate hepatic impairment. Has not been studied in patients with severe hepatic impairment.
Dilution
Available in 500-mg vials. Reconstitute each 500-mg vial by slowly directing 10 mL of NS to vial sides (50 mg/mL). Ensure wetting of entire daptomycin product. Allow vial to stand for 10 minutes, then gently rotate to ensure complete dilution. To minimize foaming, avoid vigorous agitation or shaking during or after reconstitution. Freshly reconstituted solutions range in color from pale yellow to light brown. May be administered as a 50 mg/mL reconstituted solution or may be further diluted with 50 mL of NS before administration and given as an infusion.
Filters:
No data available from manufacturer.
Storage:
Refrigerate unopened vials at 2° to 8° C (36° to 46° F). Both reconstituted and diluted solutions are stable for 12 hours at RT or up to 48 hours refrigerated. The combined time (vial and infusion bag) at RT should not exceed 12 hours. Combined refrigeration time (vial and infusion bag) should not exceed 48 hours.
Compatibility (underline indicates conflicting compatibility information)
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
Manufacturer states, “Additives or other medications should not be added to daptomycin single-use vials or infused simultaneously through the same intravenous line. If the same intravenous line is used for sequential infusion of several different drugs, the line should be flushed with a compatible infusion solution before and after infusion with daptomycin.” ■ Manufacturer states, “Daptomycin is compatible with NS and LR but is incompatible with dextrose-containing diluents.” ■ Do not use in conjunction with ReadyMED® elastomeric infusion pumps; an incompatibility occurs because of an impurity leaching from this pump system into the daptomycin solution.
One source suggests the following compatibilities:
Y-site:
Aztreonam (Azactam), caspofungin (Cancidas), ceftazidime (Fortaz), ceftriaxone (Rocephin), dopamine, doripenem (Doribax), fluconazole (Diflucan), gentamicin, heparin, levofloxacin (Levaquin), lidocaine.
Rate of administration
See Compatibility. Flushing of the IV line before and after infusion may be indicated.
Injection:
A single dose properly reconstituted and administered over 2 minutes.
Infusion:
A single dose properly diluted and administered over 30 minutes.
Actions
A cyclic lipopeptide antibacterial agent. Binds to bacterial membranes and causes a rapid depolarization of the membrane potential. Loss of the membrane potential leads to inhibition of protein, DNA, and RNA synthesis, which results in bacterial cell death. Exhibits bactericidal activity against aerobic gram-positive bacteria and has been shown to retain potency against antibiotic-resistant, gram-positive bacteria, including isolates resistant to methicillin. Cross-resistance between daptomycin and other antibacterial agents has not been reported. Highly protein bound, primarily to albumin. Site of metabolism has not been identified. Half-life is approximately 7 to 9 hours. Is excreted primarily by the kidney. A small fraction is excreted through the feces. Secreted in breast milk.
Indications and uses
Treatment of complicated skin and skin structure infections caused by susceptible strains of several aerobic gram-positive microorganisms and Staphylococcus aureus bloodstream infections (bacteremia), including those with right-sided infective endocarditis caused by methicillin-susceptible and methicillin-resistant isolates.
Limitation of use:
Daptomycin is not indicated for treatment of left-sided infective endocarditis due to S. aureus and has not been studied in patients with prosthetic valve endocarditis. ■ Not indicated for treatment of pneumonia. In Phase 3 studies of community-acquired pneumonia, the death rate and rates of serious cardiorespiratory adverse events were higher in daptomycin-treated patients than in patients treated with a comparator agent. These differences resulted from the lack of therapeutic effectiveness of daptomycin in the treatment of community-acquired pneumonia.
Contraindications
Known hypersensitivity to daptomycin.
Precautions
C/S indicated to determine susceptibility of causative organism to daptomycin. ■ To reduce the development of drug-resistant bacteria and maintain its effectiveness, daptomycin should be used to treat only those infections that are proven or strongly suspected to be caused by susceptible bacteria. ■ Combination therapy may be clinically indicated if the documented or presumed pathogens include gram-negative or anaerobic organisms. ■ Eosinophilic pneumonia has been reported. Onset is usually 2 to 4 weeks after initiation of daptomycin and improves when therapy is discontinued. Patient may present with fever, dyspnea with hypoxic respiratory insufficiency, and diffuse pulmonary infiltrates. ■ Superinfection caused by the overgrowth of nonsusceptible organisms may occur with antibiotic use. Treat as indicated. ■ Clostridium difficile–associated diarrhea (CDAD) has been reported. May range from mild diarrhea to fatal colitis. Consider in patients who present with diarrhea during or after treatment with daptomycin. ■ Skeletal muscle effects associated with daptomycin have been observed. Elevations in serum creatine phosphokinase (CPK); myopathy; and rhabdomyolysis, with or without acute renal failure, have been reported. Some cases involved patients treated concurrently with daptomycin and HMG-CoA reductase inhibitors; see Drug/Lab Interactions. ■ Cases of peripheral neuropathy have been reported. ■ Hypersensitivity reactions, including anaphylaxis, have been reported. ■ In clinical trials, decreased efficacy was observed in patients with moderate baseline renal impairment (CrCl less than 50 mL/min).
Monitor:
Obtain baseline and weekly SCr, BUN, and CPK levels. Patients who received recent, prior, or concomitant therapy with an HMG-CoA reductase inhibitor (e.g., simvastatin [Zocor], lovastatin [Mevacor]), patients with renal insufficiency, and patients who develop unexplained elevations in CPK while receiving daptomycin should be monitored more frequently. See Precautions and Antidote. ■ Monitor for S/S of hypersensitivity reactions (e.g., dyspnea, fever, flushing, hypotension, nausea, pruritus, rash, urticaria). ■ Monitor for the development of muscle pain or weakness, particularly in the distal extremities. ■ Monitor for S/S of neuropathy. ■ Repeat blood cultures indicated in patients with persisting or relapsing S. aureus infection or poor clinical response. MIC (minimum inhibitory concentration) susceptibility testing and diagnostic evaluation to rule out sequestered foci of infection may be indicated. Surgical intervention (e.g., débridement, removal of prosthetic device) and/or consideration of a change in antibacterial regimen may be required. ■ Monitor for S/S of eosinophilic pneumonia (e.g., cough, fever, difficulty breathing, shortness of breath, diffuse pulmonary infiltrates). Treatment with systemic steroids is recommended.
Patient education:
Review side effects with physician. Promptly report muscle pain or weakness, S/S of a hypersensitivity reaction (e.g., hives, rash, shortness of breath or troubled breathing, swelling of eyelids, lips, or face), S/S of neuropathy (e.g., tingling or numbness, especially in the forearm or lower leg), or new or worsening cough or fever. ■ Review medications (prescription and nonprescription) with health care provider. ■ Promptly report diarrhea or bloody stools that occur during treatment or up to several months after an antibiotic has been discontinued; may indicate CDAD and require treatment.
Maternal/child:
Category B: use during pregnancy only if clearly needed. ■ Use caution during breastfeeding. Is present in breast milk, but oral bioavailability is poor. ■ Safety and effectiveness for use in pediatric patients under 18 years of age not established. Avoid use in pediatric patients younger than 12 months of age. Animal studies suggest risk of potential effects on muscular, neuromuscular, and/or nervous systems (either peripheral and/or central).
Elderly:
Lower clinical success rates were seen in patients 65 years of age or older. In addition, adverse events were more common in this age-group. Consider age-related renal impairment. See Dose Adjustment.
Drug/lab interactions
Daptomycin does not appear to inhibit or induce the activities of the cytochrome P450 isoforms: 1A2, 2A6, 2C9, 2C19, 2D6, 2E1, and 3A4. It is unlikely that it will inhibit or induce the metabolism of drugs metabolized by this system. ■ In vitro synergistic interactions occurred with aminoglycosides, beta-lactam antibiotics, and rifampin (Rifadin) against some isolates of staphylococci and enterococci, including some methicillin-resistant Staphylococcus aureus (MRSA) isolates and some vancomycin-resistant enterococci isolates. ■ Has been administered with warfarin. Does not appear to affect the pharmacokinetics of either drug. However, daptomycin can cause a significant concentration-dependent false prolongation of PT and elevation of INR when certain recombinant thromboplastin reagents are used for the assay. This drug-lab interaction can be minimized by drawing specimens for PT/INR near the time of trough plasma concentrations of daptomycin. Evaluation of PT/INR using an alternative method may be required. ■ Inhibitors of HMG-CoA reductase (e.g., simvastatin [Zocor], atorvastatin [Lipitor]) may cause myopathy, which is manifested as muscle pain or weakness associated with elevated levels of CPK and possible rhabdomyolysis. Consider temporarily suspending the use of HMG-CoA reductase inhibitors in patients receiving daptomycin. ■ Has been used concomitantly with aztreonam (Azactam). No dose adjustment for either antibiotic was required. ■ No dose adjustment is required when given concomitantly with probenecid.
Side effects
Most side effects are mild to moderate in intensity. The most frequently reported side effects were abnormal liver function tests, dyspnea, and elevated CPK. Side effects occurring in 2% or more of patients include abdominal pain, bacteremia, chest pain, diarrhea, dizziness, edema, headache, hypertension, hypotension, insomnia, pharyngolaryngeal pain, pruritus, rash, sepsis, sweating, and urinary tract infections. Less frequently reported side effects include anxiety, back pain, Candida infections, cardiac failure, CDAD, cellulitis, confusion, cough, decreased appetite, elevated alkaline phosphatase, hyperglycemia, hypoglycemia, hypokalemia, and sore throat. Muscle pain or weakness, rhabdomyolysis (with or without renal failure), and peripheral neuropathy have been reported rarely. See Precautions. Hypersensitivity reactions including anaphylaxis, difficulty swallowing, hives, pruritus, shortness of breath, and truncal erythema have been reported. Other reactions have been reported in fewer than 1% of study patients. See manufacturer’s literature.
Post-marketing:
Angioedema, drug rash with eosinophilia and systemic symptoms (DRESS), eosinophilic pneumonia, nausea and vomiting, serious skin reactions including Stevens-Johnson syndrome, vesiculobullous rash, and visual disturbances.
Antidote
Notify physician of any side effects. Discontinue drug in patients with unexplained S/S of myopathy in conjunction with CPK elevation greater than 1,000 units/L (approximately 5 times the upper limit of normal) or in patients without reported symptoms who have marked elevation in CPK, with levels of greater than 2,000 units/L (equal to or greater than 10 times the upper limit of normal). In animal studies, skeletal muscle effects and neuropathies were reversible with discontinuation of the drug. Discontinue daptomycin at the first sign of eosinophilic pneumonia. Treatment with systemic steroids is recommended. Treat CDAD with fluids, electrolytes, protein supplements, and oral vancomycin (Vancocin) or metronidazole (Flagyl) as indicated. In severe cases, surgical evaluation may be indicated. Treat hypersensitivity reactions as indicated (e.g., oxygen, diphenhydramine, epinephrine, corticosteroids, vasopressors, and/or fluids). Approximately 15% of daptomycin is removed during a 4-hour hemodialysis run. Approximately 11% is recovered over 48 hours with peritoneal dialysis. Use of a high-flux dialysis membrane may increase the amount of drug removal. Resuscitate as necessary.
Daratumumab
(DAR-a-TOOM-ue-mab)
Darzalex
Monoclonal antibody
Antineoplastic
pH 5.5
Usual dose
Premedication:
To reduce the risk of infusion reactions, administer an IV corticosteroid (methylprednisolone 100 mg or equivalent intermediate- or long-acting corticosteroid), an oral antipyretic (acetaminophen 650 to 1,000 mg), and an oral or IV antihistamine (diphenhydramine 25 to 50 mg or equivalent) 1 hour before every infusion of daratumumab. Following the second infusion, the dose of corticosteroid may be reduced (methylprednisolone 60 mg IV).
Daratumumab:
16 mg/kg as an infusion at a specified rate; see Rate of Administration. Repeat dose weekly for Weeks 1 through 8, every 2 weeks for Weeks 9 through 24, and every 4 weeks from Week 25 onward until disease progression. If a planned dose is missed, administer as soon as possible and adjust the dosing schedule accordingly, maintaining the treatment interval.
Postinfusion medication:
To reduce the risk of delayed infusion reactions, administer an oral corticosteroid (20 mg methylprednisolone or equivalent) after all infusions on the first and second day. For patients with a history of obstructive pulmonary disorder, consider postinfusion short- and long-acting bronchodilators and inhaled corticosteroids after the first four infusions. These additional inhaled postinfusion medications may be discontinued if no major infusion reactions occur.
Herpes zoster reactivation prophylaxis:
Initiate within 1 week of starting daratumumab and continue for 3 months following treatment.
Dose adjustments
Upon resolution of Grade 1 or 2 (mild to moderate) infusion reactions, resume the infusion at no more than half the rate at which the reaction occurred. If no further reactions occur, infusion rate escalation may resume at increments and intervals as appropriate; see Rate of Administration. ■ Upon resolution to Grade 2 or lower from a Grade 3 (severe) reaction, consider restarting the infusion at no more than half the rate at which the reaction occurred. If no further reactions occur, resume rate escalation at increments and intervals as appropriate; see Rate of Administration. ■ If Grade 3 reactions occur for a second time, repeat the protocol as outlined. ■ Permanently discontinue daratumumab if a third Grade 3 or a Grade 4 (life-threatening) reaction occurs. ■ No dose adjustment required for renal impairment or mild hepatic impairment.
Dilution
Available as a 100 mg/5 mL or a 400 mg/20 mL single-dose vial (20 mg/mL). Calculate the dose (mg) based on actual body weight, total volume (mL) of daratumumab solution required, and the number of vials required using the following calculations:
Dose (mg) = Weight (kg) × Dose (mg/kg)
# of vials required = Dose (mg) ÷ 100 (400) mg/vial
# of mL required = Dose (mg) ÷ 20 mg/mL
For a 60-kg patient: [(60 kg) × (16 mg/kg)] ÷ 100 (400) mg/vial = 9.6 vials of the 100-mg/vial solution and 2.4 vials of the 400-mg/vial solution. After patient is weighed and appropriate dose is calculated, remove sufficient vials from the refrigerator. Aseptic technique imperative. Solution should be colorless to pale yellow. Do not use if opaque particles, discoloration, or other foreign particles are present. Withdraw the calculated dose (48 mL in both of the previous examples) from the required number of vials. This calculated dose must be further diluted in NS. Withdraw and discard a volume of NS equal to the calculated volume of daratumumab from a bag/container of NS based on the chart in Rate of Administration (1,000 or 500 mL) and slowly add daratumumab solution. Gently invert the bag/bottle to mix the solution. Infusion bags/containers must be made of polyvinylchloride (PVC), polypropylene (PP), polyethylene (PE), or polyolefin blend (PP+PE).
Filters:
Must be administered through an infusion set with a flow regulator and an in-line, sterile, nonpyrogenic, low–protein binding polyethersulfone (PES) filter (pore size 0.22 or 0.2 micrometer). Polyurethane (PU), polybutadiene (PBD), PVC, PP, or PE administration sets must be used.
Storage:
Refrigerate at 2° to 8° C (36° to 46° F) in original carton to protect from light. Do not freeze or shake. The diluted product may be stored for up to 24 hours if refrigerated and protected from light. Do not freeze. Bring to room temperature before infusion and use immediately. Diluted solution may develop very small, translucent to white proteinaceous particles because daratumumab is a protein. Do not use if opaque particles, discoloration, or other foreign particles are present. Infusion should be completed within 15 hours. Discard any unused product remaining in vials.
Compatibility
Manufacturer states, “Do not infuse concomitantly in the same IV line with other agents.” Compatible only with IV bags/containers of polyvinylchloride (PVC), polypropylene (PP), polyethylene (PE), or polyolefin blend (PP+PE); filters of polyethersulfone (PES); and administration sets of polyurethane (PU), polybutadiene (PBD), PVC, PP, or PE.
Rate of administration
For IV infusion only. Interrupt daratumumab infusion for infusion reactions of any severity and manage symptoms. See the following chart for daratumumab infusion rates.
| Infusion Rates for Daratumumab Administration | ||||
| Dilution Volume | Initial Rate (First Hour) | Rate Increment | Maximum Rate | |
| First infusion | 1,000 mL | 50 mL/hr | 50 mL/hr every hour | 200 mL/hr |
| Second infusion* | 500 mL | 50 mL/hr | 50 mL/hr every hour | 200 mL/hr |
| Subsequent infusions† | 500 mL | 100 mL/hr | 50 mL/hr every hour | 200 mL/hr |
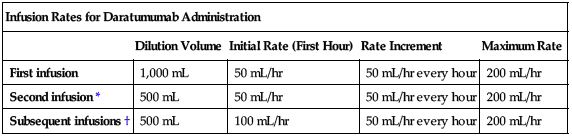
*Escalate only if there were no Grade 1 (mild) or greater infusion reactions during the first 3 hours of the first infusion.
†Escalate only if there were no Grade 1 (mild) or greater infusion reactions during a final infusion rate of equal to or greater than 100 mL/hr in the first two infusions. See Dose Adjustments.
Actions
CD38 is a transmembrane glycoprotein expressed on the surface of hematopoietic cells, including multiple myeloma and other cell types and tissues, and has multiple functions. Daratumumab is an IgG1k human monoclonal antibody that binds to CD38 and inhibits the growth of CD38-expressing tumor cells by inducing apoptosis. Steady state is achieved approximately 5 months into the every-4-week dosing period (by the 21st infusion). Half-life is approximately 18 days.
Indications and uses
Treatment of patients with multiple myeloma who have received at least 3 prior lines of therapy, including a proteasome inhibitor (PI) and an immunomodulatory agent, or who are double-refractory to a PI and an immunomodulatory agent. Accelerated approval is based on response rate; continued approval may be contingent on verification of clinical benefit.
Contraindications
Manufacturer states, “None.”
Precautions
For IV infusion only. ■ Administered under the direction of a physician knowledgeable in its use in a facility with adequate diagnostic and treatment facilities to monitor the patient and respond to any medical emergency. ■ Can cause severe infusion reactions. Approximately half of all patients experienced a reaction, most during the first infusion. Reactions can also occur with subsequent infusions. Most reactions occur during the infusion or within 4 hours of administration. ■ Interferes with serologic testing (compatibility testing, including cross-matching and antibody screening) for up to 6 months after the last infusion; see Drug/Lab Interactions. ■ Can be detected on assays used for monitoring endogenous M protein. Interference with these assays can affect the determination of complete response and of disease progression in some patients with IgG kappa myeloma protein; see Drug/Lab Interactions. ■ Has not been studied in patients with moderate to severe hepatic impairment. ■ A therapeutic protein; has the potential for immunogenicity.
Monitor:
Premedication required; see Usual Dose. ■ Type and screen patients’ blood before starting daratumumab. ■ Obtain baseline CBC and differential and monitor as indicated during therapy. ■ Monitor vital signs. ■ Monitor for S/S of infusion reactions (e.g., bronchospasm, cough, chills, dyspnea, headache, hypertension, hypotension, hypoxia, laryngeal edema, larynx and throat tightness and irritation, nasal congestion, nausea, pruritus, pulmonary edema, rash, rhinitis, urticaria, vomiting, wheezing). Immediately interrupt the infusion for infusion reactions of any grade/severity; see Dose Adjustments and Antidote.
Patient education:
Review manufacturer’s medication guide. ■ Effective contraception required for women of reproductive potential during treatment with and for 3 months after the last dose of daratumumab. ■ Immediately report any S/S of infusion-related reactions (e.g., chills, cough, difficulty breathing, headache; itchy, runny, or blocked nose). ■ Some side effects may require corticosteroid treatment and interruption or discontinuation of daratumumab. ■ Daratumumab can affect the results of some tests, including testing for complete response, and additional testing may be indicated to evaluate response to therapy. ■ Inform all health care providers, including blood transfusion centers, of daratumumab use in the event of a planned blood transfusion.
Maternal/child:
Has the potential to be transmitted from the mother to the developing fetus. Based on its mechanism of action, daratumumab may cause fetal myeloid or lymphoid-cell depletion and decreased bone density. Effective contraception required; see Patient Education. ■ Administration of live vaccines to neonates and infants exposed to daratumumab in utero should be deferred until a hematology evaluation is completed. ■ Safety for use during breast-feeding is unknown. ■ Safety and effectiveness for use in pediatric patients not established.
Elderly:
No overall differences in safety or efficacy were reported between elderly patients and younger adults.
Drug/lab interactions
No drug interaction studies have been performed. ■ Interferes with serologic testing. Daratumumab binds to CD38 on the RBCs and results in a positive indirect antiglobulin test (Coombs test). This effect may last for up to 6 months after the final daratumumab infusion. Daratumumab bound to RBCs masks detection of antibodies to minor antigens in the patient’s serum. A patient’s ABO and Rh blood type are not affected. Notify blood transfusion centers of this interference. ■ Administration of live vaccines to neonates and infants exposed to daratumumab in utero should be deferred until a hematology evaluation is completed.
Side effects
The most frequently reported side effects included back pain, cough, fatigue, fever, infusion reactions, nausea, and upper respiratory tract infections. The most frequent serious side effects were fever, general physical health deterioration, infusion reactions, and pneumonia. Treatment-emergent Grade 3 to 4 laboratory abnormalities included anemia, lymphopenia, neutropenia, and thrombocytopenia. Arthralgia, chills, constipation, decreased appetite, diarrhea, dyspnea, extremity pain, headache, herpes zoster reactivation, hypertension, musculoskeletal chest pain, nasal congestion, nasopharyngitis, and vomiting have occurred.
Antidote
Notify physician of any side effects; most will be treated symptomatically. Interrupt daratumumab infusion for infusion reactions of any severity. Permanently discontinue daratumumab if a third Grade 3 or a Grade 4 (life-threatening) hypersensitivity/infusion reaction occurs. Treat a hypersensitivity/infusion reaction with oxygen, epinephrine (Adrenalin), antihistamines (e.g., diphenhydramine [Benadryl]), vasopressors (e.g., dopamine), corticosteroids, albuterol, IV fluids, and ventilation equipment as indicated.
Darbepoetin alfa 
(DAR-beh-poh-eh-tin AL-fah)
Aranesp
Erythropoiesis-stimulating agent (ESA)
pH 6 to 6.4
Usual dose
Adult and pediatric patients:
Rate of hemoglobin increase is dose dependent and varies among patients. Availability of iron stores, baseline hemoglobin, and concurrent medical problems affect the rate and extent of response. In controlled clinical trials, patients experienced greater risks for death, serious adverse cardiovascular reactions, and stroke when administered erythropoiesis-stimulating agents (ESAs) to target a hemoglobin level of greater than 11 Gm/dL. Use the lowest dose for each patient that will gradually increase the hemoglobin concentration to avoid the need for RBC transfusion. If a patient fails to respond or maintain a response, other etiologies should be considered and evaluated. See Monitor, Precautions, and Maternal/Child.
Anemia associated with chronic kidney disease (CKD) for patients on dialysis:
Initiate darbepoetin when the hemoglobin level is less than 10 Gm/dL: Starting dose: 0.45 mcg/kg of body weight once per week or 0.75 mcg/kg once every 2 weeks as appropriate. May be given by IV or SC injection. The IV route is recommended for patients on hemodialysis; see Precautions. See Dose Adjustments and Maternal/Child.
Anemia associated with chronic kidney disease (CKD) for patients not on dialysis:
Consider initiating darbepoetin only when the hemoglobin level is less than 10 Gm/dL and the following two considerations apply: (1) the rate of hemoglobin decline indicates the likelihood of requiring a RBC transfusion, and (2) reducing the risk of alloimmunization and/or other RBC transfusion-related risks is a goal. Starting dose: 0.45 mcg/kg of body weight IV or SC given once every 4 weeks as appropriate.
Conversion from epoetin alfa to darbepoetin alfa in patients with CKD on dialysis:
Estimate starting weekly dose of darbepoetin for adult and pediatric patients based on the weekly dose of epoetin alfa at the time of substitution as shown in the following chart. Administer darbepoetin once per week in patients who were receiving epoetin alfa 2 to 3 times a week and once every 2 weeks in patients who were receiving epoetin alfa once per week. The route of administration (IV or SC) should remain the same.
| Estimated Darbepoetin Alfa Starting Dose Based on Previous Epoetin Alfa Dose for Patients with CKD on Dialysis | ||
| Previous Weekly Epoetin Alfa Dose (units/week) | Weekly Darbepoetin Alfa Dose (mcg/week) | |
| Adult | Pediatric | |
| <1,500 units/week | 6.25 mcg/week | See * |
| 1,500 to 2,499 units/week | 6.25 mcg/week | 6.25 mcg/week |
| 2,500 to 4,999 units/week | 12.5 mcg/week | 10 mcg/week |
| 5,000 to 10,999 units/week | 25 mcg/week | 20 mcg/week |
| 11,000 to 17,999 units/week | 40 mcg/week | 40 mcg/week |
| 18,000 to 33,999 units/week | 60 mcg/week | 60 mcg/week |
| 34,000 to 89,999 units/week | 100 mcg/week | 100 mcg/week |
| ≥90,000 units/week | 200 mcg/week | 200 mcg/week |
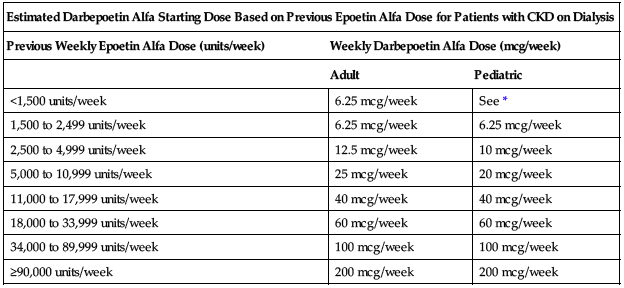
*Data insufficient to determine a darbepoetin alfa conversion dose in pediatric patients receiving a weekly epoetin alfa dose of less than 1,500 units/week.
Conversion from epoetin alfa to darbepoetin alfa in patients with CKD NOT on dialysis:
The dose conversion shown in the previous chart (for patients with CKD on dialysis) does not accurately estimate the once-monthly dose of darbepoetin.
Anemia associated with chemotherapy in cancer patients:
ESAs shortened overall survival and/or increased the risk of tumor progression or recurrence in clinical studies of patients with certain types of cancer; see Precautions. Initiate darbepoetin in patients undergoing cancer chemotherapy only if the hemoglobin is less than 10 Gm/dL and there is a minimum of 2 additional months of planned chemotherapy. Use the lowest dose necessary to avoid RBC transfusions. Starting dose: 2.25 mcg/kg of body weight SC every week until completion of chemotherapy course. An alternative schedule is 500 mcg SC every 3 weeks until completion of chemotherapy course.
Pediatric dose
Pediatric patients with CKD (less than 18 years of age):
Initiate darbepoetin when the hemoglobin level is less than 10 Gm/dL. Starting dose: 0.45 mcg/kg of body weight once per week as an IV or SC injection. Patients not receiving dialysis may also be initiated at a dose of 0.75 mcg/kg once every 2 weeks. If the hemoglobin level approaches or exceeds 12 Gm/dL, reduce or interrupt the dose of darbepoetin.
Dose adjustments
All patients with CKD:
When adjusting therapy, consider hemoglobin rate of rise, hemoglobin rate of decline, ESA responsiveness, and hemoglobin variability. A single hemoglobin excursion may not require a dose adjustment. Dose should be started slowly and adjusted for each patient to achieve and maintain the lowest hemoglobin level sufficient to avoid the need for RBC transfusion. Allow sufficient time before adjusting a dose; increased hemoglobin levels may not be observed for 2 to 6 weeks. ■ Do not increase the dose more frequently than once every 4 weeks. Decreases in dose can occur more frequently. ■ If the hemoglobin rises rapidly (e.g., more than 1 Gm/dL in any 2-week period), reduce the dose by 25% or more as needed to reduce rapid responses. ■ For patients who do not respond adequately (e.g., the hemoglobin has not increased by more than 1 Gm/dL) after 4 weeks of therapy, increase the dose by 25%. ■ For patients who do not respond adequately over a 12-week escalation period, increasing the dose further is unlikely to improve response and may increase risks. Discontinue darbepoetin if responsiveness does not improve.
Patients with CKD on dialysis:
If the hemoglobin level approaches or exceeds 11 Gm/dL, reduce or interrupt the dose of darbepoetin.
Patients with CKD NOT on dialysis:
If the hemoglobin level exceeds 10 Gm/dL, reduce or interrupt the dose of darbepoetin.
Anemia associated with chemotherapy in cancer patients:
| Dose Adjustment in Patients Undergoing Cancer Chemotherapy | ||
| Dose Adjustment | Weekly Schedule | Every-3-Week Schedule |
| If hemoglobin increases greater than 1 Gm/dL in any 2-week period or If hemoglobin reaches a level needed to avoid RBC transfusion | Reduce dose by 40% | Reduce dose by 40% |
| If hemoglobin exceeds a level needed to avoid RBC transfusion | Withhold dose until hemoglobin approaches a level at which RBC transfusions may be required Reinitiate at a dose 40% below the previous dose | Withhold dose until hemoglobin approaches a level at which RBC transfusions may be required Reinitiate at a dose 40% below the previous dose |
| If hemoglobin increases by less than 1 Gm/dL and remains below 10 Gm/dL after 6 weeks of therapy | Increase dose to 4.5 mcg/kg/week | No dose adjustment |
| If there is no response as measured by hemoglobin levels or if RBC transfusions are still required after 8 weeks of therapy Following completion of a chemotherapy course | Discontinue darbepoetin | Discontinue darbepoetin |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


