Scalp injuries: this may include contusion, abrasion, laceration, and subgaleal hematoma
Skull fractures: further classified according to the following.
Type: include linear skull fracture, comminuted skull fracture, and depressed skull fracture
Location: based on the anatomic location of the fractures such as frontal bone fracture, temporal skull fracture, and basilar skull fracture
Brain injuries: classified as follows.
Focal injuries: include contusion, laceration, and hemorrhage. The latter is further classified as epidural hematoma (EDH), subdural hematoma (SDH), subarachnoid hemorrhage, and intracerebral hematoma (ICH)
Diffuse injuries: include concussion and diffuse axonal injury (DAI)
CHART 16-1 Classification of Scalp and Craniocerebral Trauma | |
|---|---|
|

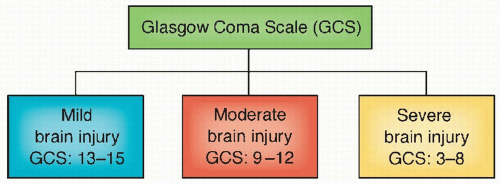 Figure 16-1 ▪ Classification of brain injury according to the Glasgow Coma Scale. |
distort tissue, alter perfusion, and result in increased intracranial pressure (ICP). Head motions caused by these mechanical forces result in contact phenomena injuries and head motions of accelerations and decelerations (Fig. 16-3).7
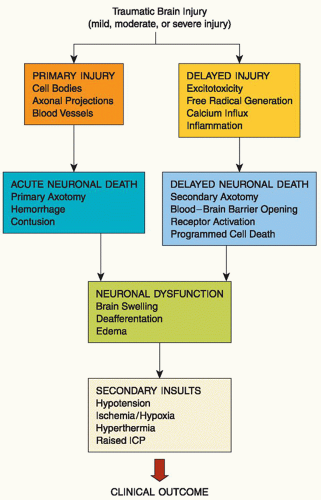 Figure 16-2 ▪ Key primary and secondary pathologic events with traumatic brain injury. ICP, intracranial pressure. (From: Dietrich, W. D. (2000). Trauma of the nervous system: Basic neuroscience of neurotrauma. In W. G. Bradley, R. B. Daroff, G. M. Fenichel, & C. D. Marsden (Eds.). Neurology in clinical practice (3rd ed., pp. 1045-1054) Boston: Butterworth & Heinemann.) |
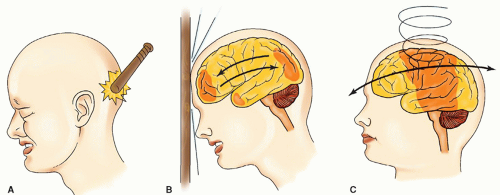 Figure 16-3 ▪ Mechanisms of injury: (A) contact, (B) acceleration-deceleration, and (C) rotational acceleration-deceleration. |
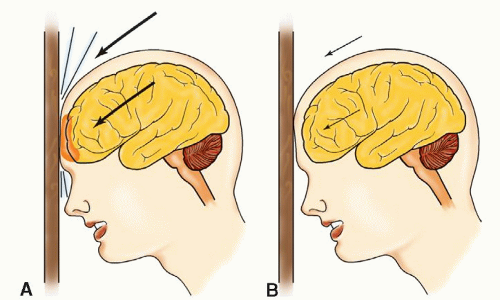 Figure 16-4 ▪ A: High-velocity contact impact on the brain and skull. B: A low-velocity contact impact damages only the skull. |
Acceleration injuries are the result of a moving object striking the head. Deceleration injuries result from the moving head striking an immobile object. The local effects of such forces are scalp laceration, skull fracture, extradural hematoma, contusions, lacerations, and intracerebral hemorrhage.8 The velocity (low or high) of the impact determines whether the injury is restricted to the scalp or skull (low velocity) or includes the brain (high velocity) (Fig. 16-4).
Acceleration-deceleration injuries are caused by abrupt changes in the velocity of the brain’s principally rapid forward movement followed by an abrupt stop (inertia) within the cranial vault. It is produced by head motion at the instant after injury and causes strain on cerebral tissue.9 These strains often operate simultaneously or in rapid succession to produce injury by compression (pushing together of tissue), tension (traction on tissue), or shearing (opposite but parallel sliding motion of the planes of an object).
where the brain tissue hits bony buttresses in the cranial vault. The effects of linear acceleration of the head are much less significant than those resulting from rotational forces. The acceleration-deceleration mechanism is responsible for two important types of injury encountered in blunt TBI, acute SDH and DAI.
TABLE 16-1 MRI FINDINGS AND DEFICITS FOR DIFFUSE AXONAL INJURY AND mTBI | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||
of EAAs is believed to be injured, energy-depleted, and depolarized neural cells (neurons and glia) that release their glutamate. Escalating levels of glutamate and aspartate stimulate specific EAA receptors that normally mediate excitatory synaptic transmission between neurons.
other hand, IL-6 demonstrates neuroprotective function by preventing TNF synthesis, promoting nerve growth factor and neuronal regeneration, and neutralizing NMDA-mediated toxicity. Another product of inflammatory cells is nitric oxide. This is synthesized by neuronal and endothelial cells and can be a potent vasodilator and thus improve perfusion, downregulate EAA, and inhibit cell death.34
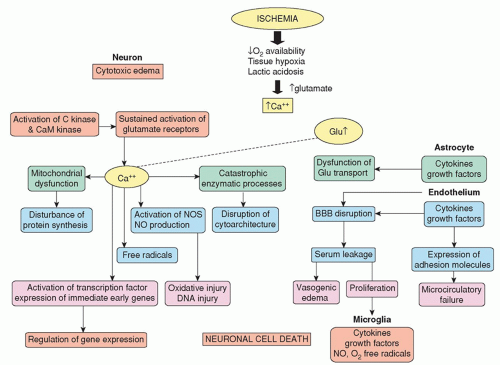 Figure 16-5 ▪ Pathophysiological changes with ischemia (need permission). |
Abrasion: the top layer of the scalp is scraped away; this is a minor injury that may cause slight bleeding. The area is cleaned and possibly dressed, and no other treatment is required.
Contusion: the scalp is bruised with possible effusion of blood into the subcutaneous layer without a break in the integrity of the skin; there is no specific treatment.
Laceration: the scalp is torn and may bleed profusely; suturing may be necessary.
Subgaleal hematoma: a hematoma in the subgaleal layer of the scalp occurs and will usually absorb on its own.
Linear: a singular fracture line occurring to the skull, which could be displaced or nondisplaced
Comminuted: the skull is splintered or shattered into pieces
Depressed: a fracture of the skull in which a fragment is depressed more than half the width of the bone; the scalp and/or dura may or may not be torn
Open depressed fracture: also known as compound skull fracture; is an opening of the skull as a result of comminuted depressed skull fractures and tearing of the dura mater and the scalp
Basal skull fracture: often arises from extension of a linear fracture into the base of the skull. The frontal and temporal bones are usually affected so that the fracture involves the anterior or middle fossa. It is important to distinguish between fractures of the cranial vault and those of the base of the skull. Although the mechanism by which the fractures arise is similar, the consequences of basilar fractures are more serious than those of cranial vault fractures. A feature of basal skull fractures is the frequency with which they traverse the paranasal air sinuses (frontal, maxillary, or ethmoid) of the frontal bone or the air sinuses located in the petrous portion of the temporal bone. The fragility of the bones in these areas and the intimate adherence of delicate dura account for the frequency of lesions in these areas and the consequent leakage of CSF through the dural tear manifested either by rhinorrhea (from the nose) or otorrhea (from the ear). Continued leakage of CSF can lead to serious infections. Such patients are at high risk for meningitis, abscess formation, and osteomyelitis from organisms gaining entry by way of the ear, nose, or paranasal sinuses through the dural tear.
Rhinorrhea (drainage of CSF, blood, or both from the nose)
Subconjunctival hemorrhage of the eye
Periorbital ecchymosis (raccoon’s eyes)
Otorrhea (drainage of CSF, blood, or both from the ear)
Hemotympanum (blood behind the tympanic membrane)
Battle’s sign (ecchymosis over mastoid bone that develops 12 to 24 hours after injury)
Conductive hearing loss (may be associated with signs of vestibular dysfunction, such as vertigo, nausea, and nystagmus)
Possible facial nerve palsy (Bell’s palsy) that appears 5 to 7 days after injury
be necessary. Surgical elevation of depressed skull fractures may be necessary with greater than 8- to 10-mm depression, neurological deficit, CSF leak, and presence or absence of open depressed fracture.35 For cosmetic and brain protective purposes, a cranioplasty with insertion of a bone or artificial graft may follow immediately or be postponed for a few months (approximately 3 to 6 months) if brain swelling is present.
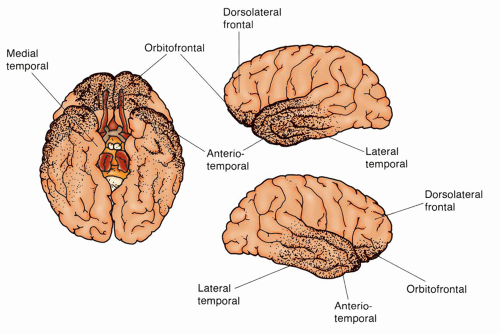 Figure 16-6 ▪ Cerebral contusions. The most frequently involved areas of the brain in cerebral contusions are the orbitofrontal and anterior-temporal regions. These are the areas that come in direct contact with the irregular bony surfaces in the inside frontal skull area. |
fracture, a penetrating wound, or an acceleration-deceleration closed injury. With acceleration-deceleration, the sites of injury are generally predictable and are located where the brain has an impact on the bony protuberances within the skull (Fig. 16-6). These areas include the frontal poles, frontal-orbital areas, frontal-temporal junction around the Sylvian fissure (where the brain is close to the lesser sphenoidal wings), and temporal poles (the inferior and lateral surfaces of the temporal lobes where there is a shelf-like separation between the anterior and middle fossae).
Fracture contusions occur at the sites of fractures and are particularly severe in the frontal lobes when associated with fractures in the anterior fossa.
Coup contusions are found directly under the areas of impact (contact).
Contrecoup contusions, by contrast, are cerebral injuries at the opposite pole of direct contact. A contrecoup injury is most often a contusion, but occasionally it may be a laceration. Both coup and contrecoup injuries are caused by the rapid acceleration-deceleration of the semisolid brain within the rigid cranial vault.
Herniation contusions occur at the time of injury at the point of contact where the medial part of the temporal lobe produces an impact at the edge of the tentorium or the cerebellar tonsils against the foramen magnum.
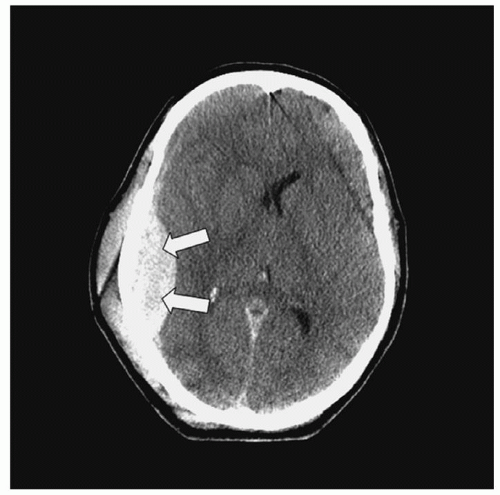 Figure 16-7 ▪ Computed tomography scan: epidural hematoma. |
Acute: up to 48 to 72 hours. This consists of clotted blood that is hyperdense and crescent shaped on CT.
Subacute: 2 to 3 days to about 2 weeks. The clot now lyses, and blood products and fluid are present. Clot will be seen isodense on CT scan.
Chronic: longer than 2 weeks to several months. The clot is a fluid mass and is hypodense on CT.
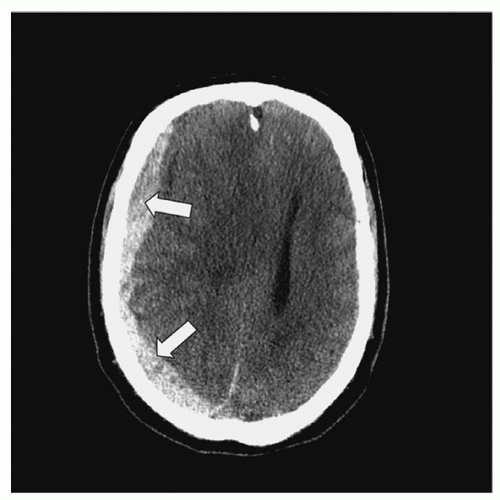 Figure 16-8 ▪ Computed tomography: subdural hematoma. |
suffered from frequent falls. Confusion was noted 2 days ago, which appeared to be worsening. Past medical history includes coronary artery disease and hypertension for which patient is taking aspirin and metoprolol, respectively. The physical examination was unremarkable except for confusion and mild lethargy. Blood chemistry, complete blood count (CBC), and drug screen were unremarkable. Head CT scan revealed an acute and chronic left frontal, parietal, and temporal SDH with 14 mm left to right midline shift. The patient was then admitted to the neurotrauma ICU for monitoring with a planned craniotomy for SDH evacuation the following day, per neurosurgery. On arrival to the ICU, 6 units of platelet transfusion was ordered for a suspected qualitative platelet dysfunction secondary to aspirin use.
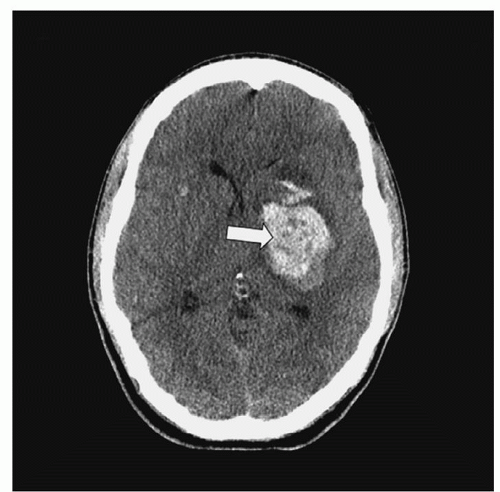 Figure 16-9 ▪ Computed tomography scan: intracerebral hematoma. |
Management consists of drainage of CSF with a ventriculostomy, ICP management, and prevention of secondary brain injuries.
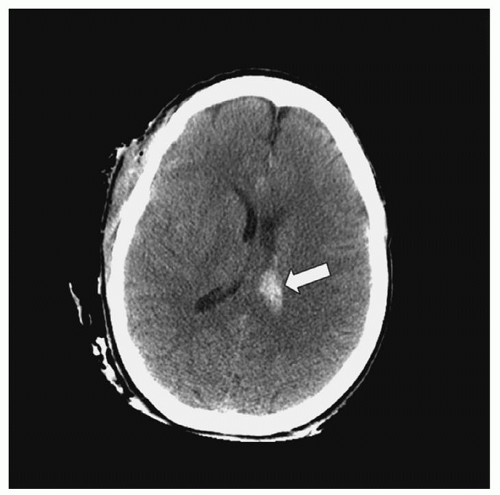 Figure 16-10 ▪ Computed tomography scan: intraventricular hematoma. |
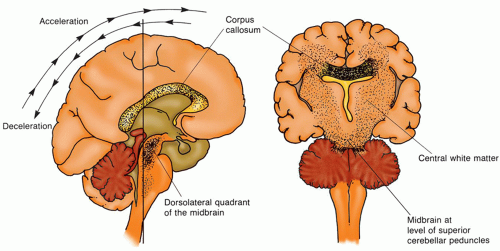 Figure 16-11 ▪ Diffuse axonal injury. Diffuse axonal injury results from acceleration-deceleration and shearing force on the brain. Depending on the severity of the injury, the areas of the brain most often affected are the corpus callosum, the dorsolateral area of the midbrain, and the parasagittal white matter. |
TABLE 16-2 CAUSES OF SECONDARY BRAIN INJURIES | ||||
|---|---|---|---|---|
|
and secondary axotomy changes due to injury.56 Table 16-3 provides a grading system DAI that occurs with acceleration-deceleration forces to the brain. The grading system is based on the distribution of pathologic findings.36
TABLE 16-3 SEQUENCE OF PRIMARY AND SECONDARY AXOTOMY CHANGES | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||
Mild DAI: coma lasting 6 to 24 hours with the patient beginning to follow commands by 24 hours. Outcome: death is uncommon, but cognitive and neurological deficits are common.
Moderate DAI: coma lasting longer than 24 hours, but without prominent brainstem signs. This is a common presentation (about 45% of all patients). Outcome: incomplete recovery in those who survive.
Severe DAI: coma is prolonged and associated with prominent brainstem signs (e.g., decortication, decerebration). This presentation is seen in about 36% of all DAI patients. Outcome: death or severe disability.
TABLE 16-4 GRADING SYSTEM FOR DIFFUSE AXONAL INJURY (DAI) BASED ON THE DISTRIBUTION OF PATHOLOGIC FINDINGS | ||
|---|---|---|
|
Depending on the severity of the concussion, a short-stay admission may be done to observe the patient’s neurological status. In other cases, a patient may be discharged home under the supervision of a responsible adult. Specific verbal and written instructions about what signs and symptoms to look for and report immediately to the physician should be provided. The patient should also be cautioned in avoiding alcohol, illicit drugs, and/or other substances such as narcotic analgesics or sedatives that may mask any signs of neurological dysfunction.62 For any persistent and/or worsening signs and symptoms, follow-up is imperative.
Tangential injuries, in which the missile does not enter the cranial cavity but produces a scalp laceration, facial injury, comminuted skull fracture, meningeal tear, or cerebral contusion-laceration.
Penetrating injuries, in which the missile enters the cranial cavity but does not pass through it, resulting in the presence of metal, bone fragments, hair, and skin within the brain. There is direct injury to cerebral tissue in the path of the bullet as well as injury to tissue from the high-pressure waves created by the high-velocity bullet. This high pressure can cause coup and contrecoup injuries, spikes in ICP, and herniation.
Through-and-through injuries, in which the missile perforates the cranial contents and leaves through an exit wound. There is one tract created from a missile entering the brain, although several tracts are possible if the bullet ricochets off bony structures within the cranial vault.
of the focus of care has been on the most severe rather than the largest group of patients the “walking wounded.”
TABLE 16-5 COMMON FACIAL FRACTURES | ||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||
in 2011 defined an mTBI as a GCS of 14 to 15, moderate as GCS of 9 to 13, and severe as 8 or less. The authors also incorporated length of coma into their definitions; severe is a coma of more than 6 hours; moderate coma as less than 6 hours or GCS never less than 8.71
TABLE 16-6 RECOMMENDATIONS FOR EVALUATION OF SPORT CONCUSSION | |||||
|---|---|---|---|---|---|
|
car). MRI was not recommended in the acute emergency setting. Patients with no isolated mTBI and a negative CT can be safely discharged from the emergency department with information about post-concussive symptoms.77 Recommendations for the evaluation of patient’s following sports concussion emphasis that MRI is more sensitive than CT at detecting axonal injury. Other techniques that may be beneficial are diffusion weighted MRI, functional MRI, MRI spectroscopy, SPECT scan or PET.78
TABLE 16-7 GRADUATED RETURN TO PLAY PROTOCOL | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


