Chapter 42 Care of Patients with Hematologic Problems
Safe and Effective Care Environment
1. Examine individual patient factors for threats to safety, especially among older adults.
2. Apply the principles of asepsis to protect immunocompromised patients.
3. Modify the environment to protect patients who have thrombocytopenia.
4. Identify appropriate community resources for the patient with a serious hematologic problem or recovering from a stem cell transplant.
5. Plan continuity of care between the hospital and community-based agencies for the patient having a stem cell transplant.
6. Verify patient identification before any form of transfusion therapy.
Health Promotion and Maintenance
7. Teach patients ways to prevent leukemia or lymphoma by avoiding known environmental causative agents.
8. Identify patients at increased risk for infection and hemorrhage.
9. Assess the patient’s endurance in performing ADLs.
10. Coordinate with a dietitian to teach patients with dietary deficiency–related anemia about the appropriate food sources for anemia prevention.
11. Teach patients and family members how to avoid injury and infection when blood counts are low.
12. Encourage the patients and family members to express their feelings about the diagnosis of a serious hematologic problem, its prognosis, and treatment.
13. Use effective communication when teaching patients and family members about what to expect during tests and therapeutic procedures.
14. Use complementary and alternative therapies along with drug therapy to improve patient comfort.
15. Identify three clinical manifestations common to patients who have any type of anemia.
16. Identify people at increased genetic risk for a hematologic disorder.
17. Prioritize nursing care for the patient who has sickle cell disease.
18. Identify the risk factors for the development of leukemia, lymphoma, and myelodysplastic syndromes.
19. Interpret laboratory data and clinical manifestations to determine the presence of infection in a patient who has neutropenia.
20. Prioritize nursing interventions for the patient with neutropenia.
21. Prioritize nursing interventions for the patient with thrombocytopenia.
22. Prioritize nursing responsibilities during transfusion therapy.
23. Identify patients at risk for complications of transfusion therapy.

http://evolve.elsevier.com/Iggy/
Answer Key for NCLEX Examination Challenges and Decision-Making Challenges
Review Questions for the NCLEX® Examination
Red Blood Cell Disorders
Anemia
There are many types and causes of anemia. Some are caused by a deficiency in one or more of the components needed to make fully functional RBCs. Such anemias can be caused by deficiencies of iron, vitamin B12, folic acid, or intrinsic factor. Other causes include a decreased rate of RBC production and increased RBC destruction. Table 42-1 lists common causes of many anemias. Despite the many causes of anemia, the effects of anemia on the patient (Chart 42-1) and the nursing care needed are similar for all types of anemia (Coyer & Lash, 2008).
TABLE 42-1 COMMON CAUSES OF ANEMIA
| TYPE OF ANEMIA | COMMON CAUSES |
|---|---|
| Sickle cell disease | |
| Glucose-6-phosphate dehydrogenase (G6PD) deficiency anemia | |
| Autoimmune hemolytic anemia | |
| Iron deficiency anemia | |
| Vitamin B12 deficiency anemia | |
| Folic acid deficiency anemia | |
| Aplastic anemia |
Anemias Resulting From Increased Destruction of Red Blood Cells
Pathophysiology
In SCD, at least 40% (and often much more) of the total hemoglobin is composed of an abnormality of the beta chains, known as hemoglobin S (HbS). HbS is sensitive to changes in the oxygen content of the RBC. When RBCs having large amounts of HbS are exposed to decreased oxygen conditions, the abnormal beta chains contract and pile together within the cell, distorting the shape of the RBC. These cells assume a sickle shape, become rigid, and clump together, causing the RBCs to become “sticky” and fragile. These clumps form masses of sickled RBCs that block blood flow (Fig. 42-1). This blood vessel obstruction, known as a vaso-occlusive event (VOE), leads to further tissue hypoxia (reduced oxygen supply) and more sickle-shaped cells, which then leads to more blood vessel obstruction and ischemia. Repeated episodes of ischemia lead to progressive organ damage from anoxia and infarction. Conditions that cause sickling include hypoxia, dehydration, infections, venous stasis, pregnancy, alcohol consumption, high altitudes, low or high environmental or body temperatures, acidosis, strenuous exercise, emotional stress, and anesthesia.
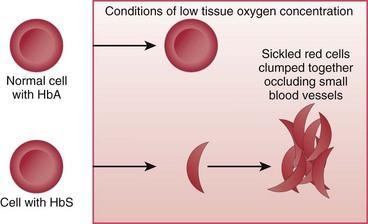
Etiology and Genetic Risk
Sickle cell disease (SCD) is a genetic disorder with an autosomal recessive pattern of inheritance (see Chapter 6). The formation of the beta chains of hemoglobin depends on a pair of gene alleles (alternate forms of a gene). A mutation in these alleles leads to the formation of HbS instead of HbA. In sickle cell disease, the patient has two HbS gene alleles, one inherited from each parent, usually resulting in 80% to 100% of the total hemoglobin being HbS. Because both hemoglobin alleles are S, sickle cell disease is sometimes also abbreviated “SS.” Patients with SCD have severe manifestations of the disease even when triggering conditions are mild. If a patient with SCD has children, each child will inherit one of the two abnormal gene alleles and at least have sickle cell trait.
In sickle cell trait, one normal gene allele and one abnormal gene allele for hemoglobin are inherited, so that only half of the hemoglobin chains produced are abnormal. Because people with this problem have one hemoglobin A allele and one hemoglobin S allele, sickle cell trait is abbreviated “AS.” The patient is a carrier of the HbS gene allele (Fig. 42-2) and can pass the trait on to his or her children. However, the patient has only mild manifestations of the disease when precipitating conditions are present because less than 40% of the hemoglobin is abnormal.
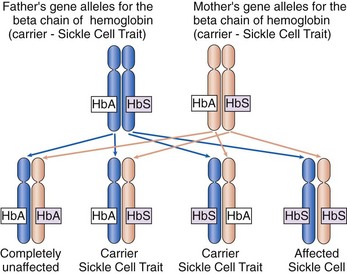
Incidence/Prevalence
Sickle cell trait and different forms of SCD occur in people of all races and ethnicities but less often among white people. In the United States, about 70,000 people have SCD, most commonly in African Americans (U.S. National Library of Medicine [USNLM], 2011). SCD occurs in 1 in 500 African Americans, and 1 in 12 to 1 in 15 (8%) African Americans are carriers of one sickle cell gene allele and have AS (USNLM, 2011).
Patient-Centered Collaborative Care
Assessment
Physical Assessment/Clinical Manifestations
Abdominal changes include major organ damage to the spleen and liver. These organs are usually the first to be damaged from many episodes of hypoxia and ischemia. In crisis, abdominal pain is diffuse and steady, involving the back and legs (Sickle Cell Information Center, 2010). Bowel sounds should be present with normal activity. Palpate the liver and spleen for enlargement. The liver or spleen may feel firm and enlarged with a nodular or “lumpy” texture in later stages of the disease. Check for guarding or rebound tenderness to palpation. A rapidly enlarging liver or spleen with increasing jaundice may indicate blood trapping in those organs.
 NCLEX Examination Challenge
NCLEX Examination ChallengeInterventions
The most common problem of SCD is pain (Granados & Jacob, 2009). The pain with sickle cell crisis is the result of tissue injury caused by poor oxygenation from obstructed blood flow. At times, patients have mild pain episodes that can be managed at home. However, pain is often severe enough to require hospitalization and large doses of opioid analgesics. Acute pain episodes have a sudden onset, usually involving the chest, back, abdomen, and extremities. Complications of SCD, such as bone necrosis, can cause severe, chronic pain, requiring large doses of opioid analgesics.
Drug therapy for patients in acute sickle cell crisis often starts with at least 48 hours of IV analgesics. (Chart 42-2 lists best practices for nursing care of the patient in sickle cell crisis.) Morphine and hydromorphone (Dilaudid) are given IV on a routine schedule or by infusion pump using patient-controlled analgesia (PCA) (see Chapter 5). Once relief is obtained, the IV dose can be tapered and the drug given orally. Avoid “as needed” (PRN) schedules because they do not provide adequate relief. Avoid IM injections because absorption is impaired by poor perfusion and sclerosed skin. Moderate pain may be treated with oral opioids or NSAIDs. (See Chapter 5 for more information on pain management.)
Chart 42-2 Best Practice for Patient Safety & Quality Care
Care of the Patient in Sickle Cell Crisis
• Administer prescribed pain medication.
• Hydrate the patient with normal saline IV and with beverages of choice (without caffeine) orally.
• Remove any constrictive clothing.
• Encourage the patient to keep extremities extended to promote venous return.
• Do not raise the knee position of the bed.
• Elevate the head of the bed no more than 30 degrees.
• Keep room temperature at or above 72° F (22.2° C).
• Avoid taking blood pressure with external cuff.
• Check circulation in extremities every hour:
Hydroxyurea (Droxia) has been successfully used to reduce the number of sickling and pain episodes (Pack-Mabien & Haynes, 2009). Hydroxyurea works by stimulating fetal hemoglobin (HbF) production. HbF is present during fetal development, but its production is turned off before birth. Increasing the level of HbF reduces sickling of red blood cells in patients with sickle cell disease. However, this drug is associated with an increased incidence of leukemia. Long-term complications should be discussed with the patient before this therapy is started. Hydroxyurea also suppresses bone marrow function, and regular follow-up to monitor complete blood counts (CBCs) for drug toxicity is important. Hydroxyurea also causes birth defects and should not be taken by anyone who is pregnant or likely to become pregnant.
 Nursing Safety Priority
Nursing Safety PriorityAction Alert
 Nursing Safety Priority
Nursing Safety PriorityPreventing Multiple Organ Dysfunction
Continued blood vessel occlusion by clumping of sickled cells increases the risk for multiple organ dysfunctions. Acute chest syndrome, in which a vaso-occlusive episode (VOE) causes infiltration and damage to the pulmonary system, is a major cause of death in adults with SCD (Hernandez & Patterson, 2009). Preventing dysfunction of any organ is important in SCD management; however, preventing heart and lung damage is a priority. Management of SCD focuses on prevention of VOEs and promotion of oxygenation.
Transfusion with RBCs can be helpful to increase HbA levels and dilute HbS levels. During transfusion, monitor the patient for complications of the procedure (discussed on pp. 900-901 in the Transfusion Reactions section).
 Decision-Making Challenge
Decision-Making ChallengeSafety; Patient-Centered Care
1. What questions related to pain would you ask this patient?
2. What drug or drugs for pain would you expect this patient to receive and by which route?
3. What diagnostic tests would you expect to be requested for this patient?
4. What nursing assessments would be most important for you to make at this time?
5. What factor or factors could have triggered this crisis episode?
Community-Based Care
Sickle cell disease (SCD) becomes worse over time. Rarely is there a true remission, although the number of crisis episodes may be reduced. Care focuses on teaching the patient and family how to prevent crises and complications (Chart 42-3). The patient with SCD may receive care in acute care, subacute care, extended or assistive care, and home care settings.
Chart 42-3 Patient and Family Education
Preparing for Self-Management: Prevention of Sickle Cell Crisis
• Drink at least 3 to 4 liters of liquids every day.
• Avoid smoking cigarettes or using tobacco in any form.
• Contact your health care provider at the first sign of illness or infection.
• Be sure to get a “flu shot” every year.
• Ask your health care provider about taking the pneumonia vaccine.
• Avoid temperature extremes of hot or cold.
• Be sure to wear socks and gloves when going outside on cold days.
• Avoid planes with unpressurized passenger cabins.
• Avoid travel to high altitudes (e.g., cities like Denver and Santa Fe).
• Ensure that any health care professional who takes care of you knows you have sickle cell disease, especially the anesthesia provider and radiologist.
• Consider genetic counseling.
• Avoid strenuous physical activities.
• Engage in mild, low-impact exercise at least three times a week when you are not in crisis.
 NCLEX Examination Challenge
NCLEX Examination ChallengePsychosocial Integrity
A. “Because you do not have the trait, you cannot have a child with SCD regardless of your partner’s sickle cell status.”
B. “Because both your parents have the trait, it is possible for you to have a child with SCD if your partner actually has the disease.”
C. “Because your brother actually has SCD, the risk for your children having SCD is 50% with each pregnancy.”
D. “Because you are a woman, your daughters will each have a 50% risk for having the disease, and all of your sons will be carriers of the trait.”
Pathophysiology
Many forms of hemolytic (blood cell–destroying) anemia are present from birth as a result of defects or deficiencies of one or more enzymes in red blood cells (RBCs). More than 200 such disorders are known. Most of these enzymes are needed to complete some critical step in RBC energy production. The most common type of inherited hemolytic anemia is the deficiency of the enzyme glucose-6-phosphate dehydrogenase (G6PD). This disease is inherited as an X-linked recessive disorder, most fully expressed in homozygous males, although partial expression (carrier state) is possible in heterozygous females. This type of anemia affects about 10% of all African Americans and also may occur in Sephardic Jews, Greeks, Iranians, Chinese, Filipinos, and Indonesians, with a frequency ranging from 5% to 40% (McCance et al., 2010).
Patient-Centered Collaborative Care
Hydration is important during an episode of hemolysis to prevent debris and hemoglobin from collecting in the kidney tubules, which can lead to acute kidney failure. Osmotic diuretics, such as mannitol (Osmitrol), may help prevent this complication. Transfusions are needed when anemia is present and kidney function is normal (see Transfusion Therapy section on pp. 897-901).
Immunohemolytic Anemia
The most common types of hemolytic anemias in North America are the immunohemolytic anemias, also referred to as autoimmune hemolytic anemias (McCance et al., 2010). The pathophysiology of hemolytic anemias involves the excessive destruction of red blood cells followed by acceleration of erythropoiesis. Acquired hemolytic syndromes result from increased RBC destruction occurring in response to trauma, viral infection, malarial infection, exposure to certain chemicals or drugs, and autoimmune reactions. All increase the rate of RBC destruction by causing membrane lysis (breakage).
In immunohemolytic anemia, immune system products (e.g., antibodies) attack a person’s own RBCs for unknown reasons. Some hemolytic anemias occur with other autoimmune disorders (e.g., systemic lupus erythematosus) or other conditions such as lymphoma, leukemia, and other neoplastic disorders (Coyer & Lash, 2008). Regardless of the cause, RBCs are viewed as non-self by the immune system and then are attacked and destroyed.
Anemias Resulting From Decreased Production of Red Blood Cells
Iron Deficiency Anemia
Adults usually have between 2 and 6 g of iron, depending on the size of the person and the amount of hemoglobin in the cells. About two thirds of this iron is contained in hemoglobin. The other one third is stored in the bone marrow, spleen, liver, and muscle. With iron deficiency, the iron stores are depleted first, followed by the hemoglobin stores. As a result, RBCs are small (microcytic) and the patient has mild symptoms of anemia, including weakness and pallor. Other clinical manifestations include fatigue, reduced exercise tolerance, and fissures at the corners of the mouth. Nails become brittle, thin, coarsely ridged, or spoon-shaped and concave (Coyer & Lash, 2008). In iron deficiency anemia, serum ferritin values are less than 10 ng/mL (normal range is 12 to 300 ng/mL).
Iron deficiency anemia is the most common anemia worldwide, especially among women, older adults, and people with poor diets. It can result from blood loss, poor GI absorption of iron, and an inadequate diet (Simmons, 2010). The problem is a decreased iron supply for the developing RBC.
Any adult with iron deficiency should be evaluated for abnormal bleeding, especially from the GI tract. Management of iron deficiency anemia involves increasing the oral intake of iron from food sources (e.g., red meat, organ meat, egg yolks, kidney beans, leafy green vegetables, and raisins). An adequate diet supplies about 10 to 15 mg of iron per day. However, only 5% to 10% of dietary iron is absorbed. This amount is enough to meet the needs of men and of women after childbearing age but is not sufficient to supply the greater needs of menstruating women. If iron losses are mild, oral iron supplements, such as ferrous sulfate, are started. This treatment should cause the hemoglobin level to rise about 2 g/dL in 4 weeks. Treatment continues until the hemoglobin level returns to normal. Instruct patients to take the iron supplement between meals for better absorption and to reduce GI distress. When iron deficiency anemia is severe, iron solutions can be given IV or IM. When given IM, these solutions must be given using the Z-track best practice method outlined in Chart 42-4. There is controversy as to whether the dorsal gluteal site or the ventrogluteal site should be used for iron dextran (Dexferrum) therapy. Although the ventrogluteal site is recommended as best practice for most IM injections, including those administered by Z-track (Zimmermann, 2010), the drug manufacturer continues to recommend the dorsal gluteal site (MD Consult, 2010). A new IV drug for iron deficiency anemia associated with chronic kidney disease is ferumoxytol (Feraheme). It has the advantage of being administered at higher dosages over a shorter period of time compared with other IV iron preparations (Belavic, 2010).
• Draw the drug up into the syringe using aseptic technique.
• Add 0.25 mL of air to the syringe.
• Discard the needle used to draw up the drug.
• Place a new needle (22-gauge, 2 to 3 inches long) on the syringe.
• Make certain that the injection site is in bright light.
• Select the dorsal gluteal site (drug manufacturer’s recommendation).
• Identify appropriate landmarks for administration into the upper, outer quadrant.
• Once the site is selected, pull the skin and subcutaneous tissues sideways away from the muscle.
• Clean the site while holding the skin and subcutaneous tissues off to the side.
• Insert the needle deeply into the muscle tissue.
• Aspirate to determine needle placement.
• Iron dextran is black; look very closely to determine whether blood is being aspirated into the syringe.
• If blood is aspirated, withdraw the needle and begin the procedure again from the first step.
• If no blood is aspirated, inject the drug slowly, followed by injection of the air bubble.
• Quickly withdraw the needle.
• Release the skin and subcutaneous tissue.
Vitamin B12 Deficiency Anemia
Vitamin B12 deficiency anemia may be mild or severe, usually develops slowly, and produces few symptoms. Patients usually have pallor and jaundice, as well as glossitis (a smooth, beefy-red tongue) (Fig. 42-3), fatigue, and weight loss. Because vitamin B12 is needed for normal nerve function, patients with pernicious anemia may also have paresthesias (abnormal sensations) in the feet and hands and poor balance (Nettina, 2009).
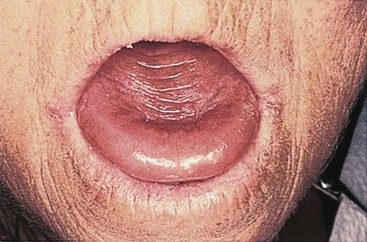
Aplastic Anemia
Pathophysiology
The most common type of the disease is acquired aplastic anemia. It can be caused by long-term exposure to toxic agents, drugs (see Table 41-3 in Chapter 41), ionizing radiation, or infection. In about 50% of cases, the cause of aplastic anemia is unknown. The disease may follow viral infection, but it is not known exactly how the bone marrow is damaged. The most common hereditary form of the disease is Fanconi’s anemia.
Patient-Centered Collaborative Care
Allogeneic hematopoietic stem cell transplantation (transplantation of bone marrow or stem cells from a sibling or matched unrelated donor) remains the preferred and most successful method of treatment for aplastic anemia that does not respond to other therapies. Cost, availability, and complications limit this treatment in aplastic anemia. For those patients who are unable to undergo such treatment or lack a suitable donor, immunosuppressive therapy remains the treatment of choice (Afable & Lyon, 2008).
Immunosuppressive therapy helps patients who have the types of aplastic anemia with a disease course similar to that of autoimmune problems. Drugs such as prednisone, antithymocyte globulin (ATG), and cyclosporine A (Sandimmune) have resulted in partial or complete remissions. The combination of ATG and cyclosporine A has been found to improve results (Afable & Lyon, 2008). Another immunosuppressant agent that has been found to improve both blood counts and transfusion requirements in patients with moderate aplastic anemia is daclizumab (Zenapax). Splenectomy (removal of the spleen) may be needed for patients with an enlarged spleen that is either destroying normal RBCs or suppressing their development.
 NCLEX Examination Challenge
NCLEX Examination ChallengeHealth Promotion and Maintenance
Polycythemia
Polycythemia Vera
Patient-Centered Collaborative Care
Polycythemia vera is a malignant disease that progresses in severity over time. If left untreated, few people with PV live longer than 2 years after diagnosis. With management by repeated pheresis (two to five times per week), the patient may live 10 to 15 years or longer. (Pheresis is the withdrawal of whole blood and removal of the patient’s RBCs to decrease the number of RBCs and reduce blood viscosity. The remaining plasma is then re-infused back into the patient.) Increasing hydration and promoting venous return help prevent clot formation. The purpose of therapy is to prevent clot formation and includes the use of anticoagulants. Chart 42-5 lists health tips for patients with PV.
Chart 42-5 Patient and Family Education
Preparing for Self-Management: Polycythemia Vera
• Drink at least 3 liters of liquids each day.
• Avoid tight or constrictive clothing, especially garters and girdles.
• Wear gloves when outdoors in temperatures lower than 50° F (10° C).
• Keep all health care–related appointments.
• Contact your physician at the first sign of infection.
• Take anticoagulants as prescribed.
• Wear support hose or stockings while you are awake and up.
• Elevate your feet whenever you are seated.
• Exercise slowly and only on the advice of your physician.
• Stop activity at the first sign of chest pain.
• Use a soft-bristled toothbrush to brush your teeth.
Myelodysplastic Syndromes
Pathophysiology
Although MDS can occur at any age, it is most common in people age 60 years or older. MDS is not officially a type of cancer but does have cancer-like features and is considered to be a precancerous state. Like cancer, MDS arises from a single population of abnormal cells. About 30% of all patients with MDS do eventually develop acute leukemia (Kumar, Abbas, Fausto, & Aster, 2010). A number of subtypes of MDS have different prognoses and responses to therapy. Patients are categorized into risk groups (i.e., low, intermediate [1 and 2], high) based on the severity of pancytopenia (low counts of all blood cell types), cytogenetic abnormalities, and numbers of blast cells (immature WBC cells) found in the bone marrow (Kurtin & Demakos, 2010).
Patient-Centered Collaborative Care
The only curative treatment for MDS is an allogeneic hematopoietic stem cell transplantation, which is often not an option because of the advanced age of the patient (Kurtin & Demakos, 2010). For low-risk and intermediate-1–risk MDS, the antitumor immunomodulatory agent lenalidomide (Revlimid) is approved for patients whose dysplastic cells have the chromosome abnormality of a deleted 5q. Two other agents that are approved for intermediate-2–risk and high-risk MDS are hypomethylating agents azacitidine (Vidaza) and decitabine (Dacogen). The results of clinical trials have increased the understanding of these treatments alone or in combination with other therapies. Effective management often requires at least 3 to 6 months to achieve a clinical response; therefore supportive care is necessary.
White Blood Cell Disorders
As discussed in Chapter 19, white blood cells (WBCs), or leukocytes, provide protection from infection and cancer development. This protection depends on maintaining normal numbers and ratios of the different mature, circulating WBCs. When any one type of WBC is present in either abnormally high or abnormally low amounts, immune function is altered to some degree, as are oxygen transport and blood clotting, placing patients at risk for many complications. This section covers the changes and nursing care for patients with disorders involving overgrowth of specific types of WBCs. (See Chapter 21 for the problems and care needs for patients with immune deficiency.)
Leukemia
Pathophysiology
Leukemias are classified by cell type. Leukemic cells coming from the lymphoid pathways (see Fig. 19-3 in Chapter 19) are typed as lymphocytic or lymphoblastic. Leukemias in which the abnormal cells come from the myeloid pathways are typed as myelocytic or myelogenous. Several subtypes exist for each of these diseases, which are classified according to the degree of maturity of the abnormal cell and the specific cell type involved. Biphenotypic leukemia is acute leukemia that shows both lymphocytic and myelocytic features.
Etiology and Genetic Risk
The exact cause of leukemia is unknown, although many genetic and environmental factors are involved in its development. The basic problem involves damage to genes controlling cell growth. This damage then changes cells from a normal to a malignant (cancer) state. Analysis of the bone marrow of a patient with acute leukemia shows abnormal chromosomes about 50% of the time (McCance et al., 2010). Possible risk factors for the development of leukemia include ionizing radiation, viral infection, exposure to chemicals and drugs, bone marrow hypoplasia (reduced production of blood cells), genetic factors, immunologic factors, environmental factors, and the interaction of these factors.
Chemicals and drugs have been linked to leukemia development because of their ability to damage DNA. Previous treatment for cancer with chemotherapy (e.g., melphalan, doxorubicin, etoposide, and cyclophosphamide) poses risks for leukemia development about 5 to 8 years after treatment. Table 41-3 in Chapter 41 lists chemicals and drugs that damage the hematologic system.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


