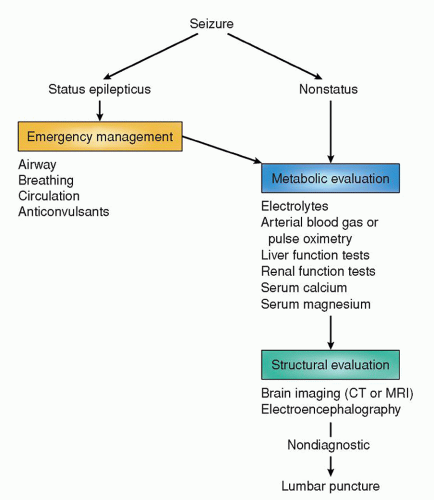
TYPE OF TUMOR |
DESCRIPTION |
LOCATION OR DEMOGRAPHIC DATA |
SIGNS AND SYMPTOMS |
TREATMENT |
PROGNOSIS |
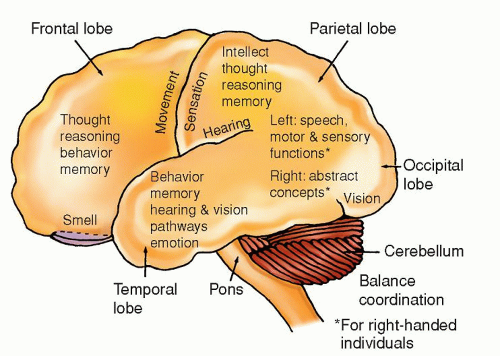
Common Brain Tumors |
Astrocytoma (grades I and II)
Constitutes 25-30% of all cerebral gliomas |
Grade I: well-defined cells
Grade II: cell differentiation less defined ↑ Cellularity |
Usually found in cerebrum, cerebellum, hypothalamus, optic nerve and chiasma, and pons
Cerebral hemisphere tumors most often found in adults 20-40 yr |
Neurological deficits depend on specific location of tumor and if it is supra- or infratentorial
Onset of a focal or generalized seizure in previously seizurefree person is most common first sign |
Surgery: gross total removal is treatment of choice, but complete removal rarely possible; partial removal may prolong life; tumor recurrence often associated with malignant progression
Radiation and Chemotherapy: controversial; not done for grade I |
5-6 yr survival on average
Range, 2-20 yr |
Anaplastic astrocytoma (grade III) |
Cellularity anaplastic: cellular atypia, ↑ mitosis |
|
|
|
15-28 mos average survival |
Glioblastoma multiforme (GBM) (also known as astrocytoma, grade IV)
Constitutes 20% of all intracranial tumors and 55% of all gliomas |
Malignant, rapidly growing
Composed of heterogeneous cells
Necrotic and hemorrhagic areas within tumor common |
Usually found in a frontal lobe
40-60 yrs most common and with increasing age
Male predilection |
Memory loss, neurobehavioral changes, seizures, speech deficits, hearing/auditory (H/A), visual deficits
Diffuse cerebral symptoms |
Surgery: resection and debulking to relieve compression and ICP
Radiation with concurrent temozolomide followed by adjuvant temozolomide |
14-16 mos average survival |
Astrocytoma of optic nerves and chiasma (spongioblastoma)
Most common in children; sometimes seen in young adults |
As the tumor grows, it enlarges the optic foramen with little distortion of surrounding structures
Slow-growing tumor |
Found along the optic nerves
Girls > boys, with 2:1 predilection |
Early symptoms include Dim vision Hemianopsia Optic atrophy
Blindness
Proptosis
Hypothalamic imbalance |
Surgery: removal possible but tumor often inaccessible
Radiation: usually poor response |
10 yrs or more |
Ependymoma (low grade and anaplastic)
Tumor of childhood and young adults |
Arises from lining of ventricles
Slow-growing |
In ventricles, particularly fourth; can attach itself to roof or floor of ventricle, or grow directly into cerebral hemisphere
Seen in children and adults up to 30 yrs, most often in men
Supratentorial more common in adults; infratentorial in children |
Rapid elevation in ICP secondary to CSF obstruction
S&S vary by location
If fourth ventricle, ↓ level of consciousness, severe H/A, VS changes with ↑ ICP, N/V, pupillary changes, hemiplegia, hemiparesthesia, seizures
If in cerebellar area, ataxia |
Surgery: removal if surgically accessible; depends on location
Radiation: for most
Chemotherapy: usually not helpful
Shunting procedure: prn to reduce ↑ ICP from obstructive hydrocephalus |
About 5-10 yrs, depending on location |
Oligodendroglioma (low grade and anaplastic) |
Calcification noted on radiologic examination in about 50% of patients |
Cerebral hemispheres, particularly frontal and temporal lobes
Found in patients 20-40 yrs |
Depends on location
Seizures are first symptoms in 50% of patients |
Surgery, chemotherapy for those with loss of heterozygosity of 1p and 19q chromosomes and radiation for those who are intact |
5-10 yrs, depending on grade |
Mixed gliomas
Named for predominant tumor cell present |
Composed histologically of two or more cell types of astrocytoma/glioblastoma, oligodendroglioma, or ependymoma in any combination |
Any place where various glioma types can be found |
Depend on location of tumor |
Depends on type of tumor
Surgery, radiation, chemotherapy |
≥5 yrs or more |
Meningioma |
Extra-axial tumor arising from dural elements
Firm, encapsulated; can erode into bone
Have estrogen and progesterone receptors; grow rapidly during pregnancy
Slow-growing; can become large before symptoms appear
Recur if not completely removed; can become malignant with reoccurrence
Compresses brain |
Predilection for areas proximal to venous sinuses
Most common in women; average age, 50 yrs
Parasagittal sinus
Lateral convexities
Sphenoid ridge
Suprasellar
Olfactory groove |
Neurological deficits caused by compression and depending on area involved
Progressive H/A, memory loss, or cognitive changes; paraparesis; seizures; urinary incontinence
Gradual development of hemiparesis, speech abnormalities; other related to area of compression
Extraocular nerve palsy, proptosis, seizures
Bitemporal hemianopsia, optic atrophy, pituitary-related hormonal imbalance
Anosmia, visual deficits, dementia, pupillary abnormalities |
Surgery: complete removal, if possible, or partial dissection
Radiation: after subtotal resection and at tumor recurrence
Immunotherapy for atypical meningiomas |
“Cure” with total removal
Many years with partial excision with radiation |
Metastatic brain tumors |
20-40% of cancer patients have metastasis to brain from other parts of the body (lungs, breast, lower GI most common)
Spread to brain by blood
Usually well differentiated from other brain tissue; lesion may be single or multiple |
Can occur anywhere
Seen as individual tumor or multiple tumors |
Depend on location H/A, paresis, and cognitive deficits most common |
Surgery: resection if possible, for singular lesion
Radiation: with multiple lesions
Gamma knife radiosurgery (for < three lesions)
Chemotherapy: similar as for primary tumor; methotrexate with oral leucovorin rescue common |
Prognosis usually based on primary cancer
1-3+yrs average |
Malignant melanomas |
Rare |
Cerebral hemispheres from a primary lesion in skin |
Depend on location |
Surgery, radiation, chemotherapy |
Few months to few years |
Primary cerebral lymphoma |
Cellular tumor
Behaves much like a glioblastoma
Occurs in adults 40-50 yrs; more common in immunocompromised patients (immunosuppressive therapy for organ transplant or people with AIDS) |
May arise in any part of brain
May be either monofocal or multifocal |
Neurocognitive and personality changes
Focal signs or ↑ ICP signs |
Biopsy followed by Decadron
Chemotherapy
Both radiation and chemotherapy are effective |
After initial response, relapse common
Average survival, 1-4 yrs |
Cerebellopontine Angle Tumors (includes several categories of tumors located in this anatomic area) |
Miscellaneous astrocytomas and meningiomas |
Can be confused with an acoustic neuroma without visualization
Definitive diagnosis made by surgical exposure, biopsy, and histologic examination |
Cerebellopontine angle |
Variation of those seen with acoustic neuroma (see below) |
Surgery: if possible; difficult surgical access (near vital centers)
Radiation: may be selected over surgery |
Depends on the type of tumor |
Acoustic neuroma (schwannoma) |
Arises from sheath of Schwann cells
Usual size: pea to walnut
Considered a benign tumor but located in an often inaccessible area
Slow-growing
Bilateral tumors are possible; when they occur, they result from a hereditary problem of chromosome 22; the tumors are part of central neurofibromatosis |
Seen most often in patients 30-49 yrs
Involves vestibular branch of CN VIII
Small tumors are confined to internal auditory canal and involve CN VIII
Large tumors extend outside internal auditory meatus
Large tumors displace CN VII and compress CN V along with CN VIII; they may also encroach on CN IX and CN X, and possibly cerebellum |
Depend on size; deficits noted on affected side
Small tumor (confined to internal auditory canal and involving CN VIII) and include: Tinnitus/vertigo
Hearing loss; most notable when using telephone or when source of sound is close to affected ear
Dizziness
Large tumor (outside auditory meatus):
S&S listed above and
Facial: loss of taste on anterior tongue, difficulty closing lower eyelid, facial weakness
Trigeminal: facial paresthesia/anesthesia, difficulty chewing
Glossopharyngeal and vagus (difficulty swallowing, hoarseness)
Cerebellar involvement (ataxia/incoordination, possibly hydrocephalus, ↑ ICP from obstruction of CSF flow secondary to displacement of pons and medulla) |
Surgery: microsurgical complete removal or debulking of larger tumors (debulking to preserve CNs involved in the tumors)
Suboccipital retrosigmoid approach for smaller tumors
Translabyrinthine approach for larger tumors
With large tumors, the tumor may entwine other CNs that would cause severe deficits if tumor were completely excised
Radiation: focused radiation (proton beam, gamma knife) alternative in older patients; scar tissue a possible problem if later surgery needed. Also used in younger patients |
Cure with small tumor and total resection; generally good outcome
Tumor regrowth possible if subtotal resection
Possible permanent hearing loss, loss of facial sensation on affected side, or facial droop
Decreased or absent corneal reflex |
Cherdoma |
Arises from embryonic remnants
May appear as a cerebellopontine angle tumor |
Predilection M > F
Occurs in patients 30-49 yrs
Found in clivus (35%) dorsum of sellae to foramen magnum and (50% in sacrococcygeal area) |
Loss of vision
Extraocular muscle paralysis
Paralyzed muscles of swallowing
Noted on MRI or CT scan |
Surgery: excision (approach varies depending on tumor location)
Radiation: conventional or proton beam |
Tumors tend to recur
Poor prognosis with aggressive and metastatic tumors |
Pituitary Tumors |
Pituitary adenomas*
Classified by type of: Hormones secreted
Effects (functioning or nonfunctioning)
Grade of sella turcica enlargement or erosion
Suprasellar extension |
Hormone(s) secreted
Prolactin (most common)
Growth hormone
ACTH
Nonfunctioning: produce S&S from compression of adjacent structures (e.g., optic nerves, bitemporal hemianopsia)
Functioning (hormonesecreting): cause endocrine syndromes (e.g., acromegaly)
Enclosed adenomas:
I—sella normal; floor may be indented
II—sella enlarged, floor intact
III—invasive adenomas; localized erosion of the floor
IV—entire floor diffusely eroded
Classified A-D by suprasellar extension
A: No suprasellar extension
B: Suprasellar bulge does not reach floor of third ventricle
C: Tumor reaches third ventricle, distorting chiasmatic recess
D: Tumor fills third ventricle almost to foramen of Munro |
Most pituitary tumors in anterior lobe
Both lobes can be damaged from compression of parasellar tumors |
In general:
Visual disorders (diminished vision with a scotoma; bitemporal hemianopsia)
Paresis of extraocular muscles
H/A
Various endocrine disorders (see below)
Abnormal sella turcica region on CT scan
Endocrine disorders:
Prolactin-secreting adenoma
Galactorrhea
Amenorrhea
Infertility
Loss of pubic hair
Impotence
↑ Serum prolactin
ACTH-secreting adenoma
Adrenal hyperplasia
Cushing’s syndrome*
Growth hormone-secreting adenoma
Giantism before puberty or closure of epiphyses
Acromegaly after puberty or closure of epiphyses (enlarged jaw, nose, tongue, hands, feet)
Thickening of soft tissue of face
Enlarged heart and pulmonary disease
Diabetes mellitus
Serum growth hormone levels >10 ng/mL
Serious complications:
Pituitary apoplexy syndrome: acute onset of ophthalmoplegia, blindness, drowsiness, and coma; death possible |
Depends on the size and type of the tumor, patient’s age, and endocrine and visual deficits; surgery, radiation, or drug therapy separately or in combination
Surgery: for smaller tumors, transsphenoidal microsurgery to remove total tumor and preserve or normalize pituitary
Radiation: conventional radiation therapy or proton beam, if available
Hormonal replacement: postsurgery, hormonal replacement possible
Other drug treatment: bromocriptine may be used to inhibit prolactin; for some patients, this is only treatment necessary for prolactinsecreting tumors |
Curable with complete resection
In others, very good outcome |
Developmental Tumors (seen sometimes in adults) |
Craniopharyngioma |
Thought to arise from Rathke’s pouch
Solid or cystic tumors
Can compress the pituitary and may even amputate the pituitary stalk
About 75% with calcified areas
Tumor growth is directed upward, resulting in invagination of the third ventricle and possible blockage of CSF flow
Optic chiasm elevated by tumor, resulting in traction on optic nerves |
In or about the sella pituitary area
Usually affects children |
Signs and symptoms of grossly ↑ ICP because of CSF flow block-age
Pituitary or hypothalamic dysfunction
Visual disturbance |
Surgery: resection by intracranial or transsphenoidal approach
Radiation: after surgery; tumor radiosensitive |
Excellent if tumor is excised with microsurgery, cure rate, 80%
Recurrence if only subtotal resection performed, even with radiation |
Epidermoid and dermoid cysts |
Cysts of congenital origin arising from the ectodermal layer; cysts lined with stratified squamous epithelium
Epidermoid cysts contain keratin, cellular debris, and cholesterol; dermoid cysts contain hair and sebaceous glands |
On bones of skull or within brain |
Depends on location |
Surgery: complete removal is usually possible |
Very good |
Genetically Related Autosomal Dominant Diseases |
Von Recklinghausen’s disease (neurofibromatosis) |
Genetic origin because of autosomal dominant mendelian trait
Skin, nervous system, bones, endocrine glands, and other organs are sites of congenital anomalies, in addition to the multiple tumors of skin
Firm, encapsulated lesions attach to the nerve |
Benign, multiple, circumscribed dermal and neural tumors with increased skin pigmentation (cosmetically offensive)
Tumors late in childhood or in early adolescence |
Spots of hyperpigmentation (café au lait) and cutaneous and subcutaneous tumors |
Surgery: possible, depending on the location of the tumor
Radiation: tumor is radioresistant |
Depends on involved area |
Hemangioblastoma (with Von Hippel-Lindau disease) |
Vascular tumor
Slow-growing |
Cerebellum (as a single or multiple lesion); less common in the medulla and cerebral hemispheres; tumor in adults |
Dizziness
Unilateral ataxia
Signs and symptoms of ↑ ICP
Possible spinal cord involvement |
Surgery: complete removal, if possible
Radiation: with recurrence |
Usually curable |
ACTH, adrenocorticotropic hormone; AIDS, acquired immunodeficiency syndrome; CN, cranial nerve; CSF, cerebrospinal fluid; CT, computed tomography; GI, gastrointestinal; H/A, headache; ICP, intracranial pressure; MRI, magnetic resonance imaging; N&V, nausea and vomiting; S&S, signs and symptoms; VS, vital signs. |
* Cushing’s syndrome comprises moon faces, “buffalo hump,” abdominal striae, pendulous abdomen; ecchymosis, hypertension, muscle weakness, osteoporosis, and high cortisol levels. |