M. Linda Workman
Assessment of the Hematologic System
Learning Outcomes
Safe and Effective Care Environment
Health Promotion and Maintenance
Psychosocial Integrity
Physiological Integrity
5 Explain the relationship between hematologic problems and the need for oxygen.
6 Describe the hematologic changes associated with aging.
7 Describe the role of platelets in hemostasis.
8 Interpret blood cell counts and clotting tests to assess hematologic status.
10 Prioritize nursing care for the patient after bone marrow aspiration.

http://evolve.elsevier.com/Iggy/
Answer Key to NCLEX Examination Challenges and Decision-Making Challenges
Audio Glossary
Key Points
Review Questions for the NCLEX® Examination
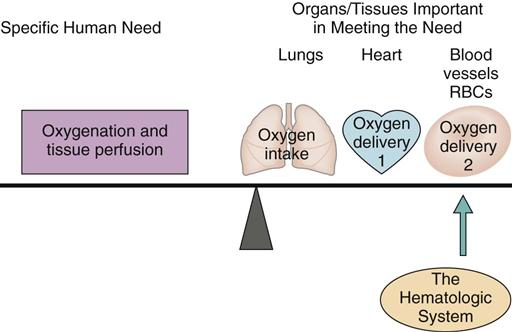
The blood, blood cells, lymph, and organs involved with blood formation or blood storage together compose the hematologic system. This system is important for oxygenation and tissue perfusion because the blood itself is one of the oxygen delivery systems (Fig. 41-1). All systems depend on the blood for oxygen perfusion. Thus any problem of the hematologic system affects total body health and well-being. This chapter, together with Chapter 19, reviews the normal physiology of the hematologic system and assessment of hematologic status.
Anatomy and Physiology Review
Bone Marrow
Bone marrow is the tissue responsible for blood formation. It produces red blood cells (RBCs, erythrocytes), white blood cells (WBCs, leukocytes), and platelets. Bone marrow also is involved in the immune responses (see Chapter 19).
Each day the bone marrow normally releases about 2.5 billion RBCs, 2.5 billion platelets, and 1 billion WBCs per kilogram of body weight. In adults, cell-producing marrow is present only in flat bones (sternum, skull, pelvic and shoulder girdles) and the ends of long bones. With aging, fatty tissue slowly replaces active bone marrow and only a small portion of the remaining marrow continues to produce blood in older adults (Touhy & Jett, 2010).
The bone marrow first produces blood stem cells, which are immature, unspecialized (undifferentiated) cells. These are capable of becoming any one of several types of blood cells: RBCs, WBCs, or platelets, depending on the body’s needs (Fig. 41-2) (McCance et al., 2010).
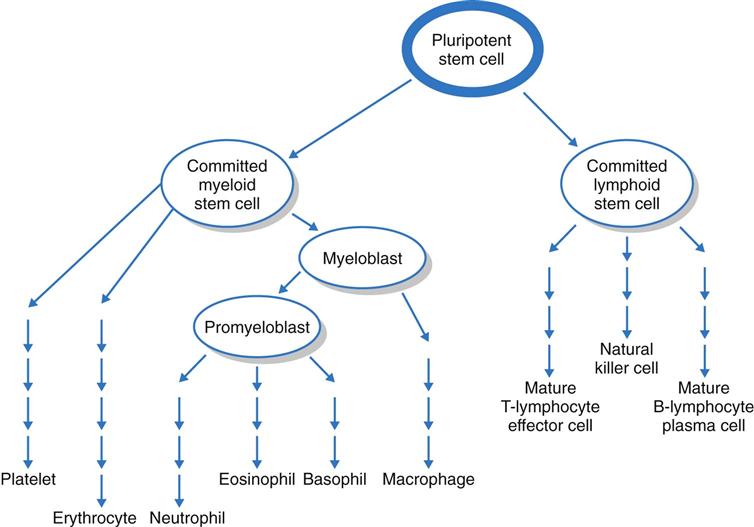
The next stage in blood cell production is the committed stem cell (also called the precursor cell). A committed stem cell enters one growth pathway and can at that point specialize (differentiate) into only one cell type. Committed stem cells actively divide but require the presence of a specific growth factor for specialization. For example, erythropoietin is a growth factor made in the kidneys that is specific for the RBC. Other growth factors influence WBC and platelet growth (see Chapters 19, 24, and 42 for discussion of growth factors and cytokines).
Blood Components
Blood is composed of plasma and cells. Plasma is part of the body’s extracellular fluid. It is similar to the interstitial fluid found between tissue cells, but plasma contains much more protein. The three major types of plasma proteins are albumin, globulins, and fibrinogen.
Albumin maintains the osmotic pressure of the blood, preventing the plasma from leaking into the tissues (see Chapter 13). Globulins have many functions, such as transporting other substances and, as antibodies, protecting the body against infection. Fibrinogen is an inactive protein that is activated to form fibrin. Fibrin molecules assemble together to form structures important in the blood clotting process.
The blood cells include RBCs, WBCs, and platelets. These cells differ in structure, site of maturation, and function.
Red blood cells (erythrocytes) are the largest proportion of blood cells. Mature RBCs have no nucleus and have a biconcave disk shape. Together with a flexible membrane, this feature allows RBCs to change their shape without breaking as they pass through narrow, winding capillaries. The number of RBCs a person has varies with gender, age, and general health, but the normal range is from 4,200,000 to 6,100,000/mm3.
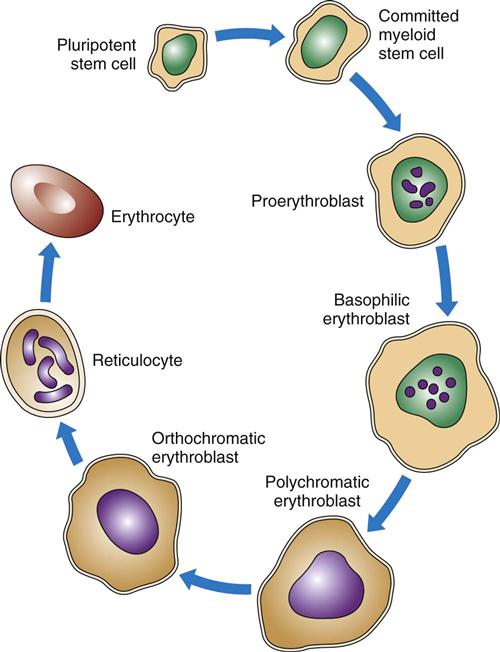
As shown in Figs. 41-2 and 41-3, RBCs start out as stem cells, enter the myeloid pathway, and progress in stages to mature RBCs (erythrocytes). Healthy, mature RBCs have a life span of about 120 days after being released into the blood. As RBCs age, their membranes become more fragile. These old cells are trapped and destroyed in the tissues, spleen, and liver. Some parts of destroyed RBCs (e.g., iron, hemoglobin) are recycled and used to make new RBCs.
The RBCs produce hemoglobin (Hgb). Each normal mature RBC contains hundreds of thousands of hemoglobin molecules. The heme part of each hemoglobin molecule needs a molecule of iron to be able to transport up to four molecules of oxygen. Therefore iron is an essential part of hemoglobin. The globin portion of hemoglobin carries carbon dioxide. RBCs also are buffers and help maintain acid-base balance.
The most important feature of hemoglobin is its ability to combine loosely with oxygen. With only a small drop in tissue oxygen levels, an increase in the transfer of oxygen from hemoglobin to tissues occurs, known as oxygen dissociation. See Chapter 29 for a complete discussion of the physiological concept of oxygen dissociation.
The total number of RBCs a person has is carefully controlled to ensure that enough are present for good oxygenation without having too many cells that could “thicken” the blood and slow its flow. Thus RBC production or erythropoiesis (selective growth of stem cells into mature erythrocytes) must be properly balanced with RBC destruction or loss. When balanced, this process helps tissue perfusion by ensuring adequate delivery of oxygen. The trigger for RBC production is an increase in the need for tissue oxygenation. The kidney produces the RBC growth factor erythropoietin at the same rate as RBC destruction or loss occurs to maintain a constant normal level of circulating RBCs. When tissue oxygenation is less than normal (hypoxia), the kidney releases more erythropoietin. This growth factor then stimulates increased RBC production in the bone marrow. When tissue oxygenation is normal or high, the kidney reduces erythropoietin levels, slowing the production of RBCs. Synthetic erythropoietin (Procrit, Epogen, EPO) has the same effect on bone marrow as the naturally occurring erythropoietin.
Many substances are needed to form hemoglobin and RBCs, including iron, vitamin B12, folic acid, copper, pyridoxine, cobalt, and nickel. A lack of any of these substances can lead to anemia. Anemia is the result of any problem that reduces the function or the number of RBCs to the point that tissue oxygen needs are not completely met.
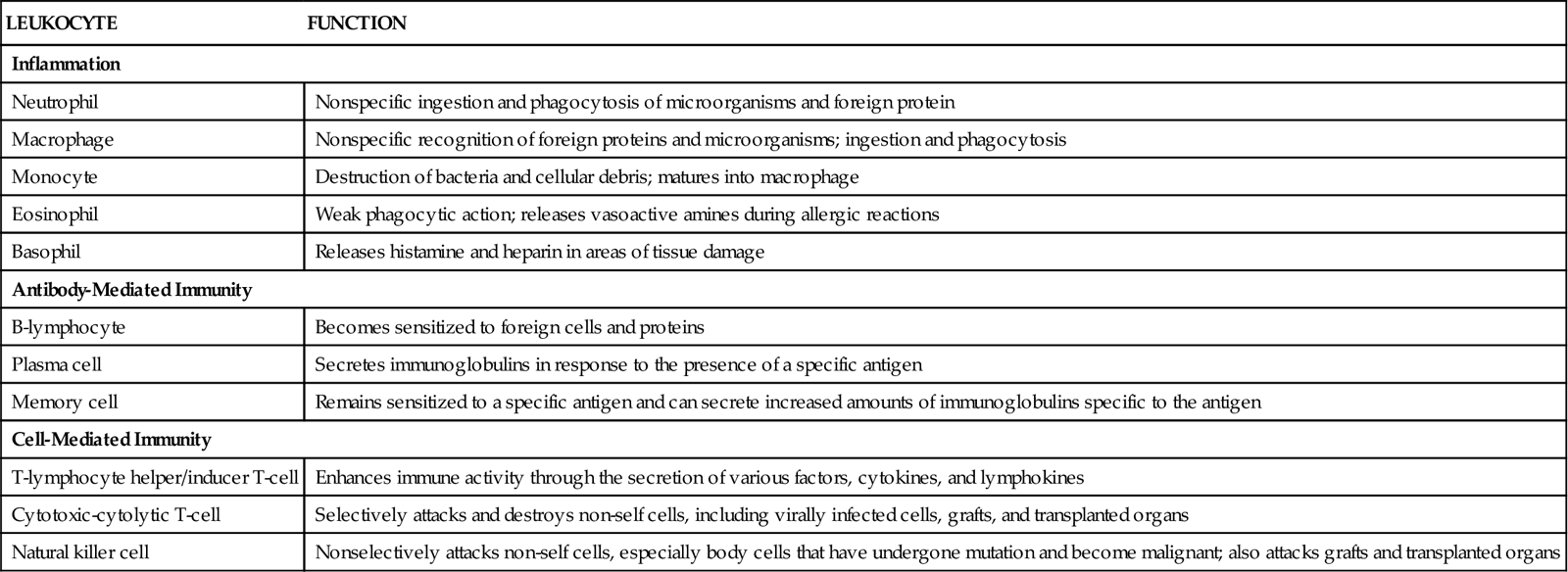
White blood cells (WBCs, leukocytes) perform actions important for protection through inflammation and immunity (Table 41-1). The many types of WBCs all have specialized functions. Many WBCs are formed in the bone marrow and are part of the hematologic system. WBC function is presented in Chapter 19.
TABLE 41-1
FUNCTIONS OF SPECIFIC LEUKOCYTES
| LEUKOCYTE | FUNCTION |
| Inflammation | |
| Neutrophil | Nonspecific ingestion and phagocytosis of microorganisms and foreign protein |
| Macrophage | Nonspecific recognition of foreign proteins and microorganisms; ingestion and phagocytosis |
| Monocyte | Destruction of bacteria and cellular debris; matures into macrophage |
| Eosinophil | Weak phagocytic action; releases vasoactive amines during allergic reactions |
| Basophil | Releases histamine and heparin in areas of tissue damage |
| Antibody-Mediated Immunity | |
| B-lymphocyte | Becomes sensitized to foreign cells and proteins |
| Plasma cell | Secretes immunoglobulins in response to the presence of a specific antigen |
| Memory cell | Remains sensitized to a specific antigen and can secrete increased amounts of immunoglobulins specific to the antigen |
| Cell-Mediated Immunity | |
| T-lymphocyte helper/inducer T-cell | Enhances immune activity through the secretion of various factors, cytokines, and lymphokines |
| Cytotoxic-cytolytic T-cell | Selectively attacks and destroys non-self cells, including virally infected cells, grafts, and transplanted organs |
| Natural killer cell | Nonselectively attacks non-self cells, especially body cells that have undergone mutation and become malignant; also attacks grafts and transplanted organs |
Platelets are the third type of blood cells. They are the smallest of the blood cells, formed as fragments of a giant precursor cell in the bone marrow, the megakaryocyte. Platelets stick to injured blood vessel walls and form platelet plugs that can stop the flow of blood from the injured site. They also produce substances important to blood clotting (coagulation), and perform most of their functions by aggregation (clumping together). Platelets help keep blood vessels intact by initiating repair after damage to small blood vessels.
Bone marrow production of platelets also is precisely controlled by the growth factor thrombopoietin. After platelets leave the bone marrow, they are stored in the spleen and then released slowly to meet the body’s needs. Normally, 80% of platelets circulate and 20% are stored in the spleen. Each platelet has a life span of 1 to 2 weeks.
Accessory Organs of Blood Formation
The spleen and liver are important accessory organs for blood production. They help regulate the growth of blood cells and form factors that ensure proper blood clotting.
The spleen is located under the diaphragm to the left of the stomach. It contains three types of tissue: white pulp, red pulp, and marginal pulp. These tissues all help balance blood cell production with blood cell destruction and assist with immunity. White pulp is filled with white blood cells (WBCs), especially lymphocytes and macrophages. It is a major site of antibody production. As whole blood filters through the white pulp, bacteria and old RBCs are removed. Red pulp contains enlarged blood vessels (sinuses) that store RBCs and platelets. Marginal pulp contains the ends of many arteries and other blood vessels.
The spleen destroys old or imperfect RBCs, breaks down the hemoglobin released from these destroyed cells, stores platelets, and filters antigens. Anyone who has had a splenectomy has reduced immune functions and is less able to remove disease-causing organisms, increasing the risk for infection and sepsis.
The liver produces prothrombin and most of the blood clotting factors. In addition, proper liver function and bile production are important in forming vitamin K in the intestinal tract. (Vitamin K is needed to produce blood clotting factors VII, IX, and X and prothrombin.) Large quantities of whole blood and blood cells can be stored in the liver. The liver also stores extra iron within the protein ferritin.
Hemostasis/Blood Clotting
Hemostasis is the multi-step process of controlled blood clotting. With proper hemostasis, localized blood clotting occurs in damaged blood vessels to prevent excessive blood loss while blood continues to circulate to all other areas. This important function is a complex process that balances blood clotting actions with anti-clotting actions. When injury occurs, hemostasis begins with the formation of a platelet plug and continues with a series of events that eventually cause the formation of a fibrin clot. Three sequential processes result in blood clotting: platelet aggregation with formation of a platelet plug, the blood clotting cascade, and the formation of a complete fibrin clot.
Platelet aggregation begins forming a platelet plug by having platelets clump together, a process essential for blood clotting. Platelets normally circulate as individual cell-like structures. They are not attracted to each other and do not clump together until activated. Activation causes platelet membranes to become sticky, allowing them to clump together. When platelets clump, they form large, semisolid plugs in blood vessel lumens and walls, disrupting local blood flow. These platelet plugs are not clots and last only a few hours. Thus they cannot provide complete hemostasis.
Substances that cause platelets to clump include adenosine diphosphate (ADP), calcium, thromboxane A2, and collagen. Platelets themselves secrete some of these substances, and other activating substances are external to the platelet. Platelet plugs start the cascade reaction that ends with local blood clotting and also are important at most steps within the cascade. When too few platelets are present, blood clotting is impaired, increasing the person’s risk for bleeding and hemorrhage.
Blood clotting is a cascade triggered by the formation of a platelet plug. The beginning of the blood clotting cascade is then rapidly amplified. The final result is much larger than the triggering event. In this sense, the cascade works like a landslide. A few small pebbles rolling down a steep hillside can dislodge large rocks and pieces of soil, causing a final enormous movement of earth. Just like landslides, cascade reactions are hard to stop once set into motion.
Intrinsic factors are conditions directly in the blood itself that first activate platelets and then trigger the blood clotting cascade (Fig. 41-4). These conditions include circulating debris and prolonged venous stasis. Continuing the cascade to the point of blood clotting requires having sufficient amounts of all the different clotting factors and cofactors (Table 41-2).
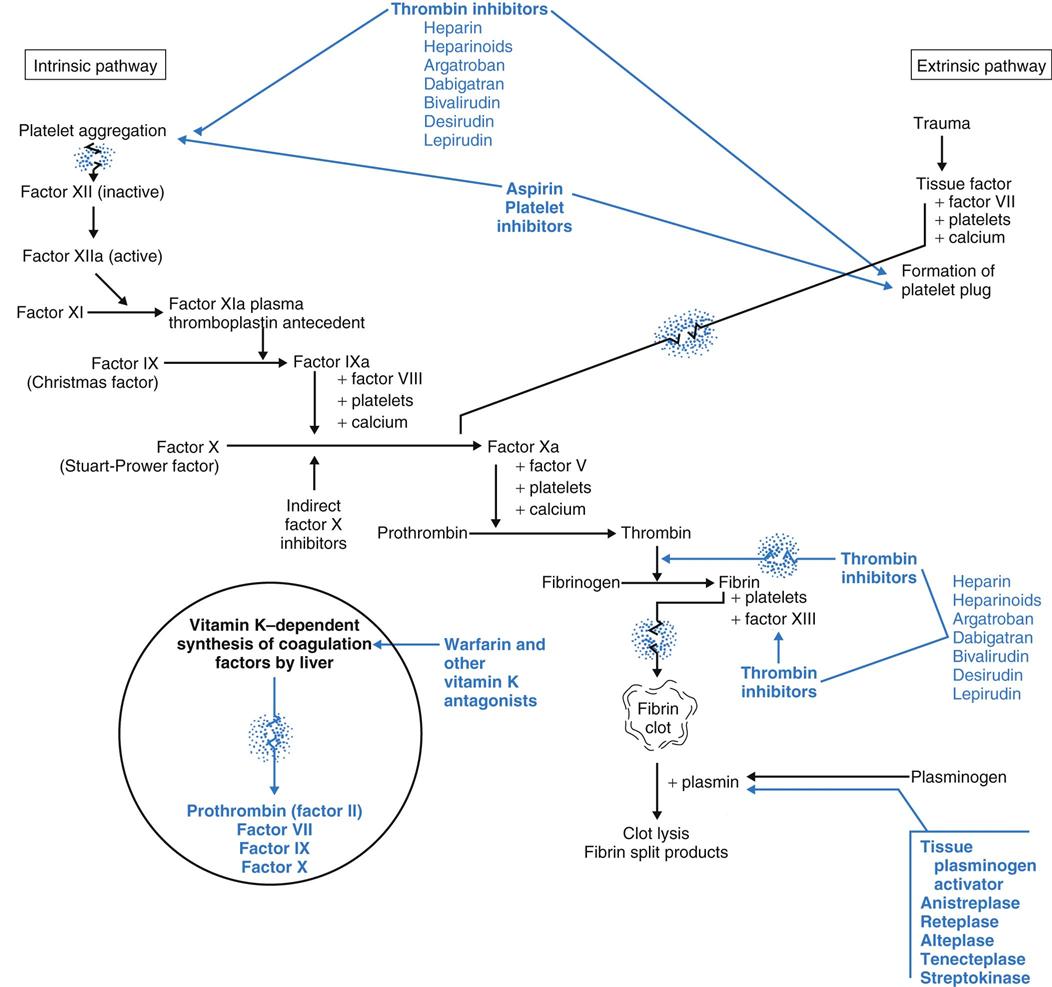
TABLE 41-2
| FACTOR | ACTION |
| I: Fibrinogen | Factor I is converted to fibrin by the enzyme thrombin. Individual fibrin molecules form fibrin threads, which are the scaffold for clot formation and wound healing. |
| II: Prothrombin | Factor II is the inactive precursor of thrombin. Prothrombin is activated to thrombin by coagulation factor X (Stuart-Prower factor). After it is activated, thrombin converts fibrinogen (coagulation factor I) into fibrin and activates factors V and VIII. Synthesis is vitamin K–dependent. |
| III: Tissue thromboplastin | Factor III interacts with factor VII to initiate the extrinsic clotting cascade. |
| IV: Calcium | Calcium (Ca2+), a divalent cation, is a cofactor for most of the enzyme-activated processes required in blood coagulation. Calcium also enhances platelet aggregation and makes red blood cells clump together. |
| V: Proaccelerin | Factor V is a cofactor for activated factor X, which is essential for converting prothrombin to thrombin. |
| VI: Discovered to be an artifact | No factor VI is involved in blood coagulation. |
| VII: Proconvertin | Factor VII activates factors IX and X, which are essential in converting prothrombin to thrombin. Synthesis is vitamin K–dependent. |
| VIII: Antihemophilic factor | Factor VIII together with activated factor IX enzymatically activates factor X. In addition, factor VIII combines with another protein (von Willebrand’s factor) to help platelets adhere to capillary walls in areas of tissue injury. A lack of factor VIII is the basis for classic hemophilia (hemophilia A). |
| IX: Plasma thromboplastin component (Christmas factor) | Factor IX, when activated, activates factor X to convert prothrombin to thrombin. This factor is essential in the common pathway between the intrinsic and extrinsic clotting cascades. A lack of factor IX causes hemophilia B. Synthesis is vitamin K–dependent. |
| X: Stuart-Prower factor | Factor X, when activated, converts prothrombin into thrombin. Synthesis is vitamin K–dependent. |
| XI: Plasma thromboplastin antecedent | Factor XI, when activated, assists in the activation of factor IX. However, a similar factor must exist in tissues. People who are deficient in factor XI have mild bleeding problems after surgery but do not bleed excessively as a result of trauma. |
| XII: Hageman factor | Factor XII is critically important in the intrinsic pathway for the activation of factor XI. |
| XIII: Fibrin-stabilizing factor | Factor XIII assists in forming cross-links among the fibrin threads to form a strong fibrin clot. |
Extrinsic factors are outside of the blood that can also activate platelets. The most common extrinsic event is trauma that damages blood vessels and exposes the platelets to collagen. Collagen then activates platelets to form a platelet plug within seconds of the trauma. The blood clotting cascade is started sooner by the extrinsic pathway because some of the steps of the intrinsic pathway are bypassed. Other blood vessel changes that can activate platelets include inflammation, bacterial toxins, or foreign proteins.
Whether the platelet plugs are formed because of abnormal blood (intrinsic factors) or by exposure to inflamed or damaged blood vessels (extrinsic factors), the end result of the cascade is the same: formation of a fibrin clot and local blood coagulation.
The steps of the cascade between the formation of a platelet plug and the formation of a fibrin clot depend on the presence of specific clotting factors. In addition, calcium and more platelets are needed at every step.
Clotting factors (see Table 41-2) are inactive enzymes that become activated in a sequence. The last part of the sequence is the activation of fibrinogen into fibrin. At each step, the activated enzyme from the previous step activates the next enzyme. The last two steps in the cascade are the activation of thrombin from prothrombin and the conversion (by thrombin) of fibrinogen into fibrin. Only fibrin molecules can begin the formation of a true clot.
Fibrin clot formation is the last phase of blood clotting. Fibrinogen is an inactive protein made in the liver. The enzyme thrombin removes the end portions of fibrinogen, converting it to active fibrin. Active fibrin molecules then link together to form fibrin threads. Fibrin threads make a netlike base to form a blood clot.
After the fibrin mesh is formed, clotting factor XIII tightens up the mesh, making it more dense and stable. More platelets stick to the threads of the mesh and attract other blood cells and proteins to form an actual blood clot. As this clot tightens (retracts), the serum is squeezed out and clot formation is complete.
 NCLEX Examination Challenge
NCLEX Examination ChallengeAnti-Clotting Forces
Because blood clotting occurs through a rapid cascade process, in theory it keeps forming fibrin clots whenever the cascade is set into motion until all blood throughout the entire body has coagulated. Such widespread clotting would lead to death. Therefore, whenever the blood clotting cascade is started, anti-clotting forces are also started to limit clot formation only to damaged areas so that normal blood flow is maintained everywhere else. When blood clotting and anti-clotting actions are normal and balanced, clotting occurs only where it is needed and normal blood flow elsewhere is maintained. The anti-clotting forces involve two types of actions. One action ensures that activated clotting factors are present only in limited amounts. The other action, fibrinolysis, prevents over-enlargement of the fibrin clot.
When the blood clotting cascade is activated, certain anti-clotting substances are also activated. Known anti-clotting proteins include protein C, protein S, and antithrombin III. Protein C and protein S increase the breakdown of clotting factors V and VIII. Antithrombin III inactivates thrombin and clotting factors IX and X. These actions prevent clots from becoming too large or forming in an area where clotting is not needed. Deficiency of any of these anti-clotting factors increases the risk for pulmonary embolism, myocardial infarction, and strokes.
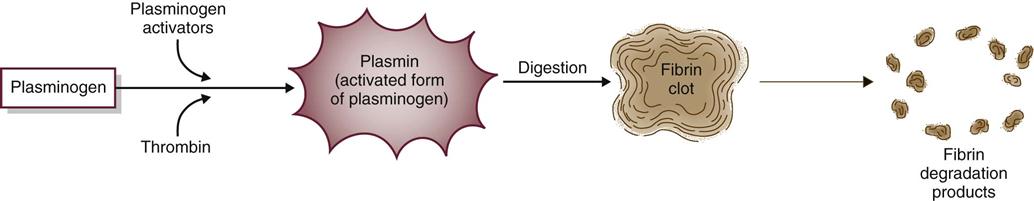
Fibrinolysis limits the size of blood clots by dissolving fibrin clot edges with special enzymes (Fig. 41-5). The process starts by activating plasminogen to plasmin. Plasmin, an enzyme, then digests fibrin, fibrinogen, and prothrombin, controlling the size of the fibrin clot.
Hematologic Changes Associated with Aging
Aging changes the cellular and plasma components of blood (Touhy & Jett, 2010). Chart 41-1 lists assessment tips for older adults. The older adult has a decreased blood volume with lower levels of plasma proteins. The lower plasma protein level may be related to a low dietary intake of proteins, as well as to reduced protein production by the older liver.
Chart 41-1
Nursing Focus on the Older Adult
Hematologic Assessment
| FINDINGS IN HEMATOLOGIC DISORDERS | NORMAL CHANGES IN THE OLDER ADULT | SIGNIFICANCE/ALTERNATIVES |
| Nail Beds (for Capillary Refill) | ||
| Pallor or cyanosis may indicate a hematologic disorder. | Thickened or discolored nails make viewing skin color beneath the nails impossible. | Use another body area, such as the lip, to assess central capillary refill. |
| Hair Distribution | ||
| Thin or absent hair on the trunk or extremities may indicate poor circulation to a particular area. | Progressive loss of body hair is a normal facet of aging. | A relatively even pattern of hair loss that has occurred over an extended period is not significant. |
| Skin Moisture | ||
| Skin dryness may indicate any of a number of hematologic disorders. | Skin dryness is a normal result of aging. | Skin moisture is not usually a reliable indicator of an underlying pathologic condition in the older adult. |
| Skin Color | ||
| Skin color changes, especially pallor and jaundice, are associated with some hematologic disorders. | Pigment loss and skin yellowing are common changes associated with aging. | Pallor in an older adult may not be a reliable indicator of anemia. Laboratory testing is required. Yellow-tinged skin in an older adult may not be a reliable indicator of increased serum bilirubin levels. Laboratory testing is required. |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


