CHAPTER 92 Systemic mycoses can be subdivided into two categories: opportunistic infections and nonopportunistic infections. The opportunistic mycoses—candidiasis, aspergillosis, cryptococcosis, and mucormycosis—are seen primarily in debilitated or immunocompromised hosts. In contrast, nonopportunistic infections can occur in any host. These latter mycoses, which are relatively uncommon, include sporotrichosis, blastomycosis, histoplasmosis, and coccidioidomycosis. Treating systemic mycoses can be difficult: These infections often resist treatment and hence may require prolonged therapy with drugs that frequently prove toxic. Drugs of choice for systemic mycoses are summarized in Table 92–1. TABLE 92–1 Drugs of Choice for Systemic Mycoses The systemic antifungal drugs fall into four classes: polyene antibiotics, azoles, echinocandins, and pyrimidine analogs. Class members and mechanisms of action are summarized in Table 92–2. TABLE 92–2 Classes of Systemic Antifungal Drugs Amphotericin B—an important but dangerous drug—is active against a broad spectrum of pathogenic fungi and is a drug of choice for most systemic mycoses (see Table 92–1). Unfortunately, amphotericin B is highly toxic: To varying degrees, infusion reactions and renal damage occur in all patients. Because of its potential for harm, amphotericin B should be employed only against infections that are progressive and potentially fatal. Amphotericin B is a drug of choice for most systemic mycoses (see Table 92–1). Before this drug became available, systemic fungal infections usually proved fatal. Treatment is prolonged; 6 to 8 weeks is common. In some cases, treatment may last for 3 or 4 months. In addition to its antifungal applications, amphotericin B is a drug of choice for leishmaniasis (see Chapter 99). Like amphotericin B, the azoles are broad-spectrum antifungal drugs. As a result, azoles represent an alternative to amphotericin B for most systemic fungal infections (see Table 92–1). In contrast to amphotericin, which is highly toxic and must be given IV, the azoles have lower toxicity and can be given by mouth. However, azoles do have one disadvantage: they inhibit hepatic cytochrome P450 drug-metabolizing enzymes, and can thereby increase the levels of many other drugs. Of the 14 azoles in current use, only 5—itraconazole, ketoconazole, fluconazole, voriconazole, and posaconazole—are indicated for systemic mycoses. Azoles used for superficial mycoses are discussed separately below. Itraconazole [Sporanox] is an alternative to amphotericin B for several systemic mycoses (see Table 92–1), and will serve as our prototype for the azole family. The drug is safer than amphotericin B and has the added advantage of oral dosing. Principal adverse effects are cardiosuppression and liver injury. Like other azoles, itraconazole can inhibit drug-metabolizing enzymes, and can thereby raise levels of other drugs. Inhibition of hepatic drug-metabolizing enzymes. Itraconazole inhibits CYP3A4 (the 3A4 isozyme of cytochrome P450) and can thereby increase levels of many other drugs (Table 92–3). The most important are cisapride, pimozide, dofetilide, and quinidine. Why? Because, when present at high levels, these drugs can cause potentially fatal ventricular dysrhythmias. Accordingly, concurrent use with itraconazole is contraindicated. Other drugs of concern include cyclosporine, digoxin, warfarin, and sulfonylurea-type oral hypoglycemics. In patients taking cyclosporine or digoxin, levels of these drugs should be monitored; in patients taking warfarin, prothrombin time should be monitored; and in patients taking sulfonylureas, levels of blood glucose should be monitored. TABLE 92–3 Some Drugs Whose Levels Can Be Increased by Azole Antifungal Drugs *In 2000, cisapride was voluntarily withdrawn from the U.S. market and is now available only through an investigational limited-access program. Fluconazole [Diflucan], a member of the azole family, is an important antifungal drug. It has the same mechanism as itraconazole: inhibition of cytochrome P450–dependent synthesis of ergosterol, with resultant damage to the cytoplasmic membrane and accumulation of ergosterol precursors. The drug is primarily fungistatic. Fluconazole is used for blastomycosis; histoplasmosis; meningitis caused by Cryptococcus neoformans and Coccidioides immitis; and vaginal, oropharyngeal, esophageal, and disseminated Candida infections. In addition, fluconazole is used investigationally for leishmaniasis (see Chapter 99). • To ensure that voriconazole levels are adequate, voriconazole should not be combined with powerful P450 inducers, including rifampin, rifabutin, carbamazepine, and phenobarbital. • To avoid excessive voriconazole levels, voriconazole should not be combined with powerful P450 inhibitors. • To avoid toxicity from accumulation of other drugs, voriconazole should not be combined with some agents that are P450 substrates, including cisapride, pimozide, and sirolimus. Ketoconazole is an alternative to amphotericin B for systemic mycoses. The drug is much less toxic than amphotericin and only somewhat less effective. Specific indications are listed in Table 92–1. Responses to ketoconazole are slow. Accordingly, the drug is less useful for severe, acute infections than for long-term suppression of chronic infections. Ketoconazole is also a valuable drug for superficial mycoses.
Antifungal agents
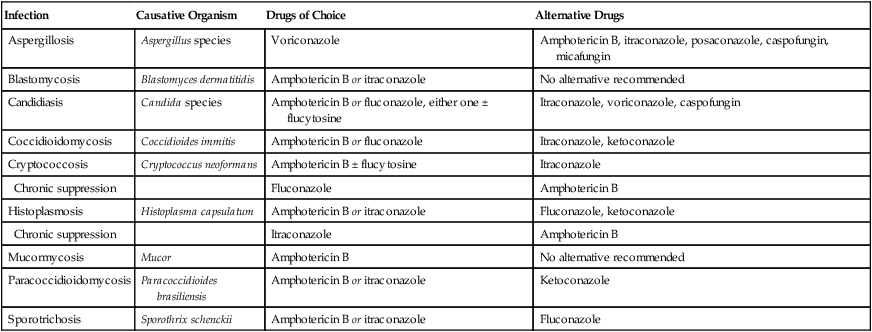
Drugs for systemic mycoses

Infection
Causative Organism
Drugs of Choice
Alternative Drugs
Aspergillosis
Aspergillus species
Voriconazole
Amphotericin B, itraconazole, posaconazole, caspofungin, micafungin
Blastomycosis
Blastomyces dermatitidis
Amphotericin B or itraconazole
No alternative recommended
Candidiasis
Candida species
Amphotericin B or fluconazole, either one ± flucytosine
Itraconazole, voriconazole, caspofungin
Coccidioidomycosis
Coccidioides immitis
Amphotericin B or fluconazole
Itraconazole, ketoconazole
Cryptococcosis
Cryptococcus neoformans
Amphotericin B ± flucytosine
Itraconazole
Chronic suppression
Fluconazole
Amphotericin B
Histoplasmosis
Histoplasma capsulatum
Amphotericin B or itraconazole
Fluconazole, ketoconazole
Chronic suppression
Itraconazole
Amphotericin B
Mucormycosis
Mucor
Amphotericin B
No alternative recommended
Paracoccidioidomycosis
Paracoccidioides brasiliensis
Amphotericin B or itraconazole
Ketoconazole
Sporotrichosis
Sporothrix schenckii
Amphotericin B or itraconazole
Fluconazole

Drug Class
Mechanism of Action
Class Members
Polyene Antibiotics
Bind to ergosterol and thereby disrupt the fungal cell membrane
Amphotericin B
Azoles
Inhibit synthesis of ergosterol and thereby disrupt the fungal cell membrane
Fluconazole
Itraconazole
Ketoconazole
Posaconazole
Voriconazole
Echinocandins
Inhibit synthesis of beta-1,3-d-glucan and thereby disrupt the fungal cell wall
Anidulafungin
Caspofungin
Micafungin
Pyrimidine Analogs
Disrupt synthesis of RNA and DNA
Flucytosine
Amphotericin B, a polyene antibiotic
Therapeutic uses
Azoles
Itraconazole
Drug interactions.

Target Drug
Class
Consequence of Excessive Level
Pimozide [Orap]
Antipsychotic
Fatal dysrhythmias
Dofetilide [Tikosyn]
Antidysrhythmic
Fatal dysrhythmias
Quinidine
Antidysrhythmic
Fatal dysrhythmias
Cisapride [Propulsid]*
Prokinetic agent
Fatal dysrhythmias
Warfarin [Coumadin]
Anticoagulant
Bleeding
Sulfonylureas
Oral hypoglycemic
Hypoglycemia
Phenytoin [Dilantin]
Antiseizure drug
Central nervous system toxicity
Cyclosporine [Sandimmune]
Immunosuppressant
Increased nephrotoxicity
Tacrolimus [Prograf]
Immunosuppressant
Increased nephrotoxicity
Lovastatin [Mevacor]
Antihyperlipidemic
Rhabdomyolysis
Simvastatin [Zocor]
Antihyperlipidemic
Rhabdomyolysis
Eletriptan [Relpax]
Antimigraine
Coronary vasospasm
Fentanyl [Duragesic, others]
Opioid analgesic
Fatal respiratory depression
Calcium channel blockers
Antihypertensive, antianginal
Cardiosuppression
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree



Antifungal agents
 ] belongs to a drug class known as polyene antibiotics, so named because their structures contain a series of conjugated double bonds. Nystatin, another antifungal drug, is in the same family.
] belongs to a drug class known as polyene antibiotics, so named because their structures contain a series of conjugated double bonds. Nystatin, another antifungal drug, is in the same family.
Get Clinical Tree app for offline access