CHAPTER 49 There are two basic types of dysrhythmias: tachydysrhythmias (dysrhythmias in which heart rate is increased) and bradydysrhythmias (dysrhythmias in which heart rate is slowed). In this chapter, we only consider the tachydysrhythmias. This is by far the largest group of dysrhythmias and the group that responds best to drugs. We do not discuss the bradydysrhythmias because they are few in number and are commonly treated with electronic pacing. When drugs are indicated, atropine (see Chapter 14) and isoproterenol (see Chapter 17) are usually the agents of choice. For the heart to pump effectively, contraction of the atria and ventricles must be coordinated. Coordination is achieved through precise timing and routing of impulse conduction. In the healthy heart, impulses originate in the sinoatrial (SA) node, spread rapidly through the atria, pass slowly through the atrioventricular (AV) node, and then spread rapidly through the ventricles via the His-Purkinje system (Fig. 49–1). The fibers of the His-Purkinje system consist of specialized conducting tissue. The function of these fibers is to conduct electrical excitation very rapidly to all parts of the ventricles. Stimulation of the His-Purkinje system is caused by impulses leaving the AV node. These impulses are conducted rapidly down the bundle of His, enter the right and left bundle branches, and then distribute to the many fine branches of the Purkinje fibers (see Fig. 49–1). Because impulses travel quickly through this system, all regions of the ventricles are stimulated almost simultaneously, producing synchronized ventricular contraction with resultant forceful ejection of blood. Profiles of fast and slow potentials are depicted in Figure 49–2. Please note that action potentials in this figure represent the electrical activity of single cardiac cells. Such single-cell recordings, which are made using experimental preparations, should not be confused with the ECG, which is made using surface electrodes, and hence reflects the electrical activity of the entire heart. As indicated in panel A of Figure 49–2, fast potentials have five distinct phases, labeled 0, 1, 2, 3, and 4. As we discuss each phase, we focus on its ionic basis and its relationship to the actions of antidysrhythmic drugs. During phase 4, two types of electrical activity are possible: (1) the membrane potential may remain stable (solid line in Fig. 49–2A), or (2) the membrane may undergo spontaneous depolarization (dashed line). In cells undergoing spontaneous depolarization, the membrane potential gradually rises until a threshold potential is reached. At this point, rapid phase 0 depolarization takes place, setting off a new action potential. Hence, it is phase 4 depolarization that gives cardiac cells automaticity (ie, the ability to initiate an action potential through self-excitation). The capacity for self-excitation makes potential pacemakers of all cells that have it. Slow potentials occur in cells of the SA node and AV node. The profile of a slow potential is depicted in Figure 49–2B. Like fast potentials, slow potentials are generated by ion fluxes. However, the specific ions involved are not the same for every phase. Phase 0 (depolarization phase) of slow potentials differs significantly from phase 0 of fast potentials. As we can see from Figure 49–2, whereas phase 0 of fast potentials is caused by a rapid influx of sodium, phase 0 of slow potentials is caused by slow influx of calcium. Because calcium influx is slow, the rate of depolarization is slow; and because depolarization is slow, these potentials conduct slowly. This explains why impulse conduction through the AV node is delayed. Phase 0 of the slow potential is of therapeutic significance in that drugs that suppress calcium influx during phase 0 can slow (or stop) AV conduction. The major components of an ECG are illustrated in Figure 49–3. As we can see, three features are especially prominent: the P wave, the QRS complex, and the T wave. The P wave is caused by depolarization in the atria. Hence, the P wave corresponds to atrial contraction. The QRS complex is caused by depolarization of the ventricles. Hence, the QRS complex corresponds to ventricular contraction. If conduction through the ventricles is slowed, the QRS complex will widen. The T wave is caused by repolarization of the ventricles. Hence, this wave is not associated with overt physical activity of the heart. The mechanism for establishing a reentrant circuit is depicted in Figure 49–4, panels A and B. In the figure, the inverted Y-shaped structure represents a branched Purkinje fiber terminating on a strip of ventricular muscle, which appears as a horizontal bar. Normal impulse conduction is shown in Figure 49–4A. As indicated by the arrows, impulses travel down both branches of the Purkinje fiber to cause excitation of the muscle at two locations. Impulses created within the muscle travel in both directions (to the right and left) away from their sites of origin. Those impulses that are moving toward each other meet midway between the two branches of the Purkinje fiber. Since in the wake of both impulses the muscle is in a refractory state, neither impulse can proceed further, and hence both impulses stop. Figure 49–4B depicts a reentrant circuit. The shaded area in branch 1 of the Purkinje fiber represents a region of one-way conduction block. This region prevents conduction of impulses downward (toward the muscle) but does not prevent impulses from traveling upward. (Impulses can travel back up the block because impulses in muscle are very strong, and hence are able to pass the block, whereas impulses in the Purkinje fiber are weaker, and hence are unable to pass.) A region of one-way block is essential for reentrant activation. There are two mechanisms by which drugs can abolish a reentrant dysrhythmia. First, drugs can improve conduction in the sick branch of the Purkinje fiber, and can thereby eliminate the one-way block (Fig. 49–4C). Alternatively, drugs can suppress conduction in the sick branch, thereby converting one-way block into two-way block (Fig. 49–4D). According to the Vaughan Williams classification scheme, the antidysrhythmic drugs fall into five groups (Table 49–1). As the table shows, there are four major classes of antidysrhythmic drugs (classes I, II, III, and IV) and a fifth group that includes adenosine and digoxin. Membership in classes I through IV is determined by effects on ion movements during slow and fast potentials (see Fig. 49–2). TABLE 49–1 Vaughan Williams Classification of Antidysrhythmic Drugs Class II consists of beta-adrenergic blocking agents. As suggested by Figure 49–2, these drugs reduce calcium entry (during fast and slow potentials) and they depress phase 4 depolarization (in slow potentials only). Beta blockers have three prominent effects on the heart: Cardiac effects of the beta blockers are nearly identical to those of the calcium channel blockers. Only two calcium channel blockers—verapamil and diltiazem—are employed as antidysrhythmics. As indicated in Figure 49–2, calcium channel blockade has the same impact on cardiac action potentials as does beta blockade. Accordingly, verapamil, diltiazem, and beta blockers have nearly identical effects on cardiac function—namely, reduction of automaticity in the SA node, delay of conduction through the AV node, and reduction of myocardial contractility. Antidysrhythmic benefits derive from suppressing AV nodal conduction. Of the mechanisms by which drugs can cause dysrhythmias, one deserves special mention: prolongation of the QT interval. As discussed in Chapter 7, drugs that prolong the QT interval increase the risk of torsades de pointes, a dysrhythmia that can progress to fatal ventricular fibrillation. All class IA and class III agents cause QT prolongation, and hence must be used with special caution. With proper treatment, digoxin-induced dysrhythmias can almost always be controlled. Treatment is discussed at length in Chapter 48. If antidysrhythmic drugs are required, lidocaine and phenytoin are the agents of choice. In patients with digoxin toxicity, DC cardioversion may bring on ventricular fibrillation. Accordingly, this procedure should be used only when absolutely required. Torsades de pointes is an atypical, rapid, undulating ventricular tachydysrhythmia that can evolve into potentially fatal ventricular fibrillation. The main factor associated with development of torsades de pointes is prolongation of the QT interval, which can be caused by a variety of drugs (see Table 7–2 in Chapter 7), including class IA and class III antidysrhythmic agents. Acute management consists of IV magnesium plus cardioversion for sustained ventricular tachycardia. Several measures can help minimize risk. These include • Starting with low doses and increasing them gradually. • Using a Holter monitor during initial therapy to detect danger signs—especially QT prolongation, which can precede torsades de pointes. • Monitoring plasma drug levels. Unfortunately, although drug levels can be good predictors of noncardiac toxicity (eg, quinidine-induced nausea), they are less helpful for predicting adverse cardiac effects. As discussed above, the antidysrhythmic drugs fall into four main groups—classes I, II, III, and IV—plus a fifth group that includes adenosine and digoxin. The pharmacology of these drugs is presented below, and summarized in Table 49–2. TABLE 49–2 Properties of Antidysrhythmic Drugs
Antidysrhythmic drugs
Introduction to cardiac electrophysiology, dysrhythmias, and the antidysrhythmic drugs
Electrical properties of the heart
Impulse conduction: pathways and timing
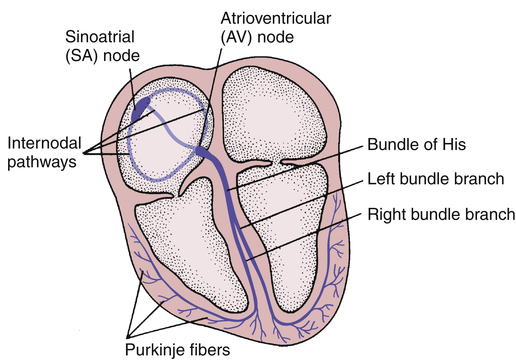
 Cardiac conduction pathways.
Cardiac conduction pathways.
His-Purkinje system.
Cardiac action potentials
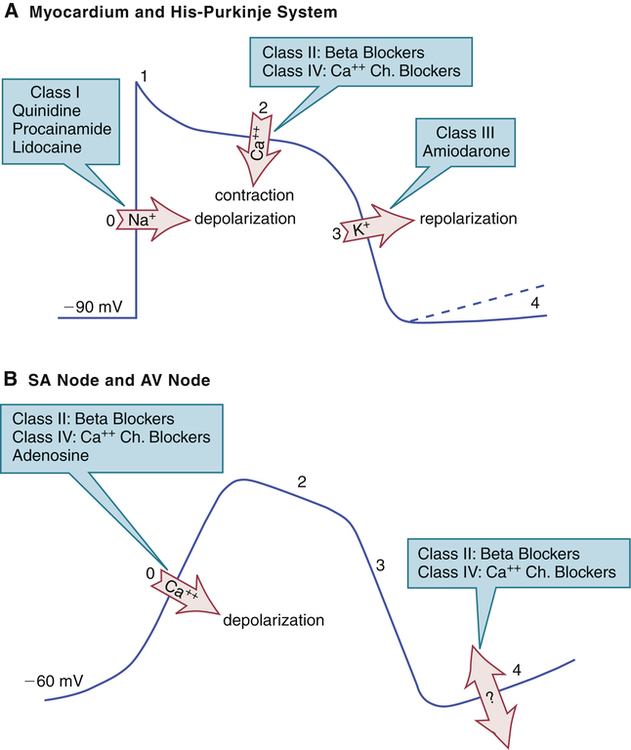
 Ion fluxes during cardiac action potentials and effects of antidysrhythmic drugs.
Ion fluxes during cardiac action potentials and effects of antidysrhythmic drugs.
A, Fast potential of the His-Purkinje system and atrial and ventricular myocardium. Blockade of sodium influx by class I drugs slows conduction in the His-Purkinje system. Blockade of calcium influx by beta blockers and calcium channel blockers decreases contractility. Blockade of potassium efflux by class III drugs delays repolarization and thereby prolongs the effective refractory period. B, Slow potential of the sinoatrial (SA) node and atrioventricular (AV) node. Blockade of calcium influx by beta blockers, calcium channel blockers, and adenosine slows AV conduction. Beta blockers and calcium channel blockers decrease SA nodal automaticity (phase 4 depolarization); the ionic basis of this effect is not understood.
Fast potentials
Phase 4.
Slow potentials
Phase 0.
The electrocardiogram
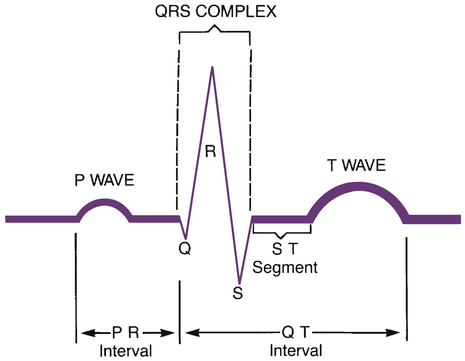
 The electrocardiogram.
The electrocardiogram.
Generation of dysrhythmias
Disturbances of conduction
Reentry (recirculating activation).
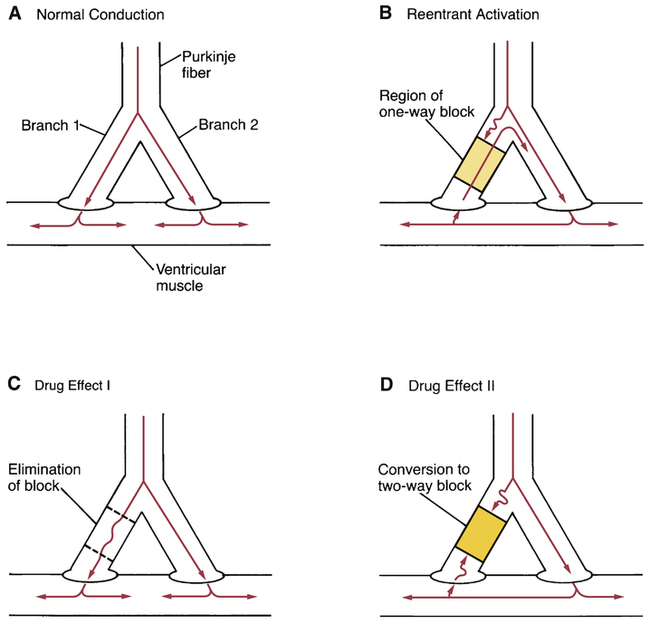
 Reentrant activation: mechanism and drug effects.
Reentrant activation: mechanism and drug effects.
A, In normal conduction, impulses from the branched Purkinje fiber stimulate the strip of ventricular muscle in two places. Within the muscle, waves of excitation spread from both points of excitation, meet between the Purkinje fibers, and cease further travel. B, In the presence of one-way block, the strip of muscle is excited at only one location. Impulses spreading from this area meet no impulses coming from the left and, therefore, can travel far enough to stimulate branch 1 of the Purkinje fiber. This stimulation passes back up the fiber, past the region of one-way block, and then stimulates branch 2, causing reentrant activation. C, Elimination of reentry by a drug that improves conduction in the sick branch of the Purkinje fiber. D, Elimination of reentry by a drug that further suppresses conduction in the sick branch, thereby converting one-way block into two-way block.
Classification of antidysrhythmic drugs

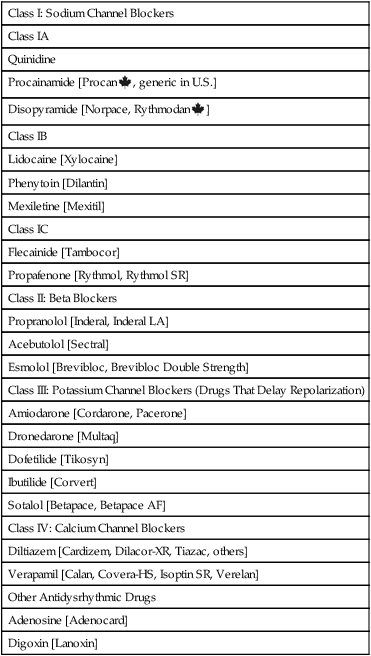
Class I: Sodium Channel Blockers
Class IA
Quinidine
Procainamide [Procan  , generic in U.S.]
, generic in U.S.]
Disopyramide [Norpace, Rythmodan  ]
]
Class IB
Lidocaine [Xylocaine]
Phenytoin [Dilantin]
Mexiletine [Mexitil]
Class IC
Flecainide [Tambocor]
Propafenone [Rythmol, Rythmol SR]
Class II: Beta Blockers
Propranolol [Inderal, Inderal LA]
Acebutolol [Sectral]
Esmolol [Brevibloc, Brevibloc Double Strength]
Class III: Potassium Channel Blockers (Drugs That Delay Repolarization)
Amiodarone [Cordarone, Pacerone]
Dronedarone [Multaq]
Dofetilide [Tikosyn]
Ibutilide [Corvert]
Sotalol [Betapace, Betapace AF]
Class IV: Calcium Channel Blockers
Diltiazem [Cardizem, Dilacor-XR, Tiazac, others]
Verapamil [Calan, Covera-HS, Isoptin SR, Verelan]
Other Antidysrhythmic Drugs
Adenosine [Adenocard]
Digoxin [Lanoxin]
Class II: beta blockers
Class IV: calcium channel blockers
Prodysrhythmic effects of antidysrhythmic drugs
Overview of common dysrhythmias and their treatment
Ventricular dysrhythmias
Digoxin-induced ventricular dysrhythmias.
Torsades de pointes.
Principles of antidysrhythmic drug therapy
Minimizing risks
Pharmacology of the antidysrhythmic drugs

Drug
Usual Route
Effects on the ECG
Major Antidysrhythmic Applications
Class IA
Quinidine
PO
Widens QRS, prolongs QT
Broad spectrum: used for long-term suppression of ventricular and supraventricular dysrhythmias
Procainamide
PO
Widens QRS, prolongs QT
Broad spectrum: similar to quinidine, but toxicity makes it less desirable for long-term use
Disopyramide
PO
Widens QRS, prolongs QT
Ventricular dysrhythmias
Class IB
Lidocaine
IV
No significant change
Ventricular dysrhythmias
Mexiletine
PO
No significant change
Ventricular dysrhythmias
Phenytoin
PO
No significant change
Digoxin-induced ventricular dysrhythmias
Class IC
Flecainide
PO
Widens QRS, prolongs PR
Maintenance therapy of supraventricular dysrhythmias
Propafenone
PO
Widens QRS, prolongs PR
Maintenance therapy of supraventricular dysrhythmias
Class II
Propranolol
PO
Prolongs PR, bradycardia
Dysrhythmias caused by excessive sympathetic activity; control of ventricular rate in patients with supraventricular tachydysrhythmias
Acebutolol
PO
Prolongs PR, bradycardia
Premature ventricular beats
Esmolol
IV
Prolongs PR, bradycardia
Control of ventricular rate in patients with supraventricular tachydysrhythmias
Class III
Amiodarone
PO
Prolongs QT and PR, widens QRS
Life-threatening ventricular dysrhythmias, atrial fibrillation*
Dronedarone
PO
Prolongs QT and PR, widens QRS
Atrial flutter, atrial fibrillation
Sotalol
IV
Prolongs QT and PR, bradycardia
Life-threatening ventricular dysrhythmias, atrial fibrillation/flutter
Dofetilide
PO
Prolongs QT
Highly symptomatic atrial dysrhythmias
Ibutilide
IV
Prolongs QT
Atrial flutter, atrial fibrillation
Class IV
Verapamil
PO
Prolongs PR, bradycardia
Control of ventricular rate in patients with supraventricular tachydysrhythmias
Diltiazem
IV
Prolongs PR, bradycardia
Same as verapamil
Others
Adenosine
IV
Prolongs PR
Termination of paroxysmal supraventricular tachycardia
Digoxin
PO
Prolongs PR, depresses ST
Control of ventricular rate in patients with supraventricular tachydysrhythmias ![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Antidysrhythmic drugs
Only gold members can continue reading. Log In or Register to continue
