CHAPTER 103 In this chapter, we continue our discussion of anticancer agents, focusing on two large groups of drugs: hormonal agents and targeted drugs. The hormonal agents, used primarily for breast cancer and prostate cancer, mimic or suppress the actions of endogenous hormones. The so-called targeted drugs bind with specific molecular targets on cancer cells, and thereby suppress tumor growth and promote cell death. Unlike the cytotoxic agents discussed in Chapter 102, many of which are cell-cycle phase specific, the drugs addressed here lack phase specificity. In addition, many of these drugs lack the serious toxicities associated with cytotoxic agents, including bone marrow suppression, stomatitis, alopecia, and severe nausea and vomiting. Nonetheless, most of these have severe toxicities of their own. Breast cancer is second only to skin cancer as the most common cancer among women in the United States. In 2011, an estimated 230,480 new cases were diagnosed and 39,520 were fatal. Between 2002 and 2003, the incidence of breast cancer dropped by 7%. Why? Because many women stopped using menopausal hormone therapy (formerly known as hormone replacement therapy) after data from the Women’s Health Initiative showed that it increased risk of breast cancer and heart disease (see Chapter 61). Not only has the incidence of breast cancer declined, so has the death rate, thanks to earlier detection and improved treatment. Principal treatment modalities are surgery, radiation, cytotoxic drugs (chemotherapy), and hormonal drugs. Surgery and radiation are considered primary therapy; chemotherapy and hormonal therapy are used as adjuvants. For a woman with early breast cancer, treatment typically consists of surgery (using total mastectomy or partial mastectomy [lumpectomy]) followed by local radiation. After that, chemotherapy is used to kill cells left behind after surgery and radiation, and to kill cells that may have metastasized to other sites. Finally, hormonal agents are taken for several years to reduce recurrence. Increasingly, chemotherapy is used before surgery—so-called neoadjuvant therapy—to shrink large tumors, and thereby permit lumpectomy in women who would otherwise require mastectomy. Drugs for adjuvant therapy are summarized in Table 103–1. TABLE 103–1 Drugs for Adjuvant Therapy of Breast Cancer *Denosumab is also available as Prolia for treating postmenopausal osteoporosis. †Zoledronate is also available as Reclast for treating osteoporosis and Paget’s disease. What about breast cancer prevention? Currently, two drugs are approved for preventing breast cancer in women at high risk. Both drugs are selective estrogen receptor modulators, or SERMS. One of the drugs—raloxifene [Evista]—is approved only for postmenopausal women. The other drug—tamoxifen [Nolvadex]—is approved for premenopausal and postmenopausal women. In clinical trials, these drugs reduced the risk of breast cancer by about 50%. However, they both pose a risk of thrombosis, and tamoxifen also poses a risk of endometrial cancer. Nonetheless, in women at high risk for breast cancer, the benefits of these drugs outweigh the risks. Raloxifene is discussed in Chapter 75. Tamoxifen is discussed below. Two other drugs—exemestane [Aromasin] (discussed below) and lasofoxifene [Oporia]—can also prevent breast cancer, but are not yet approved for this use. • Ages 40 through 49—Tamoxifen is a good choice, except for women at risk for thrombosis. • Ages 50 through 59—Tamoxifen is a good choice, except for women at risk for thrombosis and for women who still have a uterus (the risk of endometrial cancer is higher for women over age 50). • Age 60 and above—Tamoxifen is not recommended (although older women have the highest risk for breast cancer, they also have the highest risk for complications from tamoxifen). Despite this recommendation, tamoxifen is effective in older women, and hence some authorities would still recommend using it, but only after the woman carefully considers the benefit versus the risks. To help determine who is at high risk for breast cancer, the National Cancer Institute has created an Internet-based Breast Cancer Risk Assessment Tool. You can access the tool at www.cancer.gov/bcrisktool. Cytotoxic drugs may be used before breast surgery or after. When used before surgery, chemotherapy can shrink large tumors, thereby permitting lumpectomy in women who would otherwise require a mastectomy. When used after surgery, chemotherapy can kill cancer cells that remain in the breast, as well as cells that may have metastasized to distant sites. A common regimen for breast cancer consists of doxorubicin (an anthracycline-type anticancer antibiotic) plus cyclophosphamide (an alkylating agent) followed by paclitaxel (a mitotic inhibitor). Representative cytotoxic drugs are presented in Table 103-1. Women with breast cancer are at risk for skeletal-related events (SREs), especially hypercalcemia and fractures. There are two causes: the cancer itself and the drugs used for treatment. In breast cancer, most metastases occur in bone. These metastases promote hypercalcemia by increasing the activity of osteoclasts, the cells that promote bone resorption. Not only does resorption promote hypercalcemia, it weakens bone, and thereby increases the risk of fractures. Fracture risk is further increased by use of antiestrogens and aromatase inhibitors. Why? As we discussed in Chapter 61, estrogens promote bone health by inhibiting bone resorption and promoting bone deposition. Hence, by removing the influence of estrogen, the antiestrogens and aromatase inhibitors accelerate bone resorption and reduce bone deposition. Both actions weaken bone, and thereby increase the risk of fractures. To reduce the risk of SREs, we can treat patients with denosumab or a bisphosphonate (usually zoledronate). Denosumab, marketed as Xgeva, is indicated for preventing (delaying) SREs in patients with breast cancer and other solid tumors that have metastasized to bone. Benefits derive from inhibiting the formation and function of osteoclasts. Efficacy was demonstrated in three double-blind trials that compared denosumab with zoledronate. One trial enrolled patients with breast cancer, one enrolled patients with prostate cancer, and one enrolled patients with other cancers, including multiple myeloma, kidney cancer, small cell lung cancer, and non-small cell lung cancer. Patients received either denosumab (120 mg subQ every 4 weeks) or zoledronate (4 mg IV every 4 weeks). The results? In patients with breast cancer or prostate cancer, denosumab was superior to zoledronate at delaying SREs. In patients with other cancers, denosumab was equal to zoledronate at delaying SREs. Principal adverse effects of denosumab are hypocalcemia, serious infections, skin reactions, and osteonecrosis of the jaw. The pharmacology of denosumab is presented in Chapter 75. Cancer of the prostate is the most common cancer among men in the United States. In 2011, an estimated 240,890 new cases were diagnosed, and 33,720 were fatal. For men with localized prostate cancer, the preferred treatments are surgery and radiation, with or without adjunctive use of drugs. For men with metastatic prostate cancer, drug therapy and castration are the only options. Among the drugs employed, agents for androgen deprivation therapy (ADT) comprise the largest and most widely used group. The only other choices are cytotoxic drugs and a new immunotherapy known as sipuleucel-T [Provenge]. As with breast cancer, most metastases (65% to 75%) go to bone. To minimize hypercalcemia and fractures caused by bone metastases, men may take zoledronate [Zometa] or denosumab [Xgeva] (see discussion of breast cancer above). The drugs used to treat prostate cancer are summarized in Table 103–2. TABLE 103–2 *Gonadotropin-releasing hormone agonists, also known as luteinizing hormone–releasing hormone (LHRH) agonists. †Denosumab is also available as Prolia for treating postmenopausal osteoporosis. ‡Zoledronate is also available as Reclast for treating osteoporosis and Paget’s disease. The gonadotropin-releasing hormone (GnRH) agonists suppress production of androgens by the testes—but not by the adrenals and prostate cancer cells. Currently, four GnRH agonists are available: leuprolide, triptorelin, goserelin, and histrelin. All four are indicated for cancer of the prostate. In addition, leuprolide is used for endometriosis (see Chapter 63). Ketoconazole [Nizoral], used primarily for fungal infections (see Chapter 92), can be used off-label for prostate cancer. As with abiraterone, benefits derive from inhibiting testicular, adrenal, and prostatic production of androgens. Ketoconazole is employed as secondary therapy in men who have rising prostate-specific antigen levels despite treatment with a GnRH agonist plus an antiandrogen. Dosages are higher than those used for antifungal therapy (400 mg 3 times a day compared with 200 mg once a day), and hence side effects are common. Among these are nausea, vomiting, fatigue, skin changes, liver damage, and gynecomastia. Because high-dose ketoconazole can suppress adrenal production of glucocorticoids, the drug is usually combined with hydrocortisone (to avoid adrenal insufficiency).
Anticancer drugs II: hormonal agents, targeted drugs, and other noncytotoxic anticancer drugs
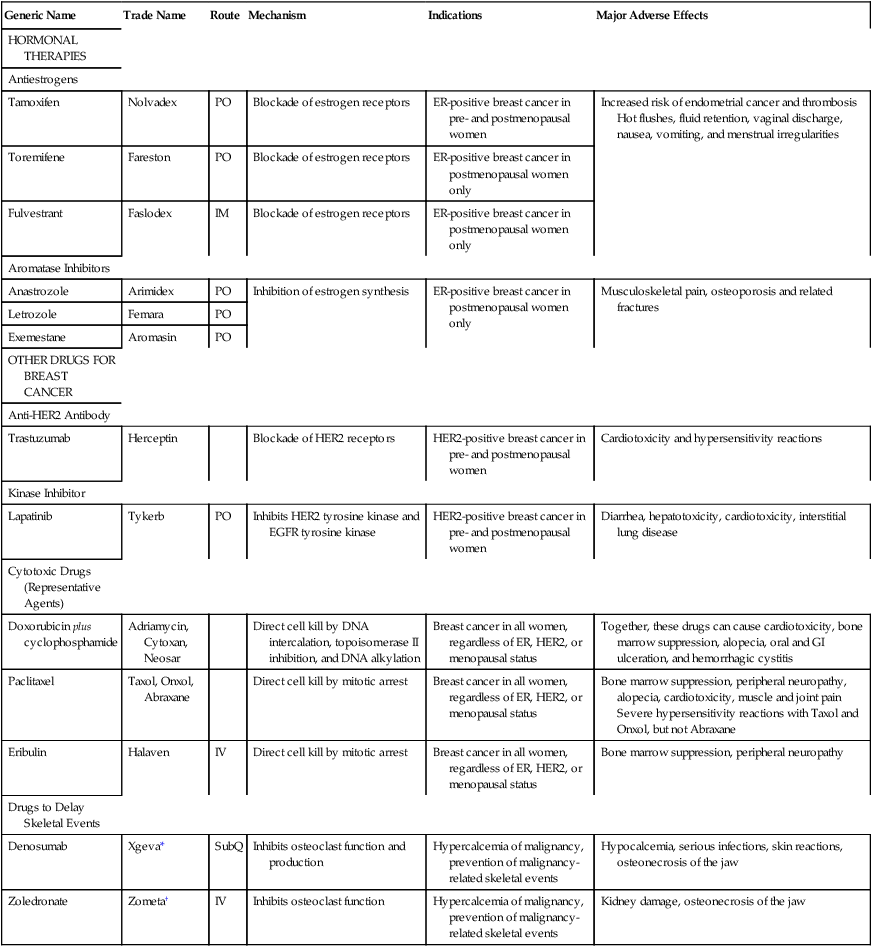
Drugs for breast cancer

Generic Name
Trade Name
Route
Mechanism
Indications
Major Adverse Effects
HORMONAL THERAPIES
Antiestrogens
Tamoxifen
Nolvadex
PO
Blockade of estrogen receptors
ER-positive breast cancer in pre- and postmenopausal women
Increased risk of endometrial cancer and thrombosis
Hot flushes, fluid retention, vaginal discharge, nausea, vomiting, and menstrual irregularities
Toremifene
Fareston
PO
Blockade of estrogen receptors
ER-positive breast cancer in postmenopausal women only
Fulvestrant
Faslodex
IM
Blockade of estrogen receptors
ER-positive breast cancer in postmenopausal women only
Aromatase Inhibitors
Anastrozole
Arimidex
PO
Inhibition of estrogen synthesis
ER-positive breast cancer in postmenopausal women only
Musculoskeletal pain, osteoporosis and related fractures
Letrozole
Femara
PO
Exemestane
Aromasin
PO
OTHER DRUGS FOR BREAST CANCER
Anti-HER2 Antibody
Trastuzumab
Herceptin
Blockade of HER2 receptors
HER2-positive breast cancer in pre- and postmenopausal women
Cardiotoxicity and hypersensitivity reactions
Kinase Inhibitor
Lapatinib
Tykerb
PO
Inhibits HER2 tyrosine kinase and EGFR tyrosine kinase
HER2-positive breast cancer in pre- and postmenopausal women
Diarrhea, hepatotoxicity, cardiotoxicity, interstitial lung disease
Cytotoxic Drugs (Representative Agents)
Doxorubicin plus cyclophosphamide
Adriamycin, Cytoxan, Neosar
Direct cell kill by DNA intercalation, topoisomerase II inhibition, and DNA alkylation
Breast cancer in all women, regardless of ER, HER2, or menopausal status
Together, these drugs can cause cardiotoxicity, bone marrow suppression, alopecia, oral and GI ulceration, and hemorrhagic cystitis
Paclitaxel
Taxol, Onxol, Abraxane
Direct cell kill by mitotic arrest
Breast cancer in all women, regardless of ER, HER2, or menopausal status
Bone marrow suppression, peripheral neuropathy, alopecia, cardiotoxicity, muscle and joint pain
Severe hypersensitivity reactions with Taxol and Onxol, but not Abraxane
Eribulin
Halaven
IV
Direct cell kill by mitotic arrest
Breast cancer in all women, regardless of ER, HER2, or menopausal status
Bone marrow suppression, peripheral neuropathy
Drugs to Delay Skeletal Events
Denosumab
Xgeva*
SubQ
Inhibits osteoclast function and production
Hypercalcemia of malignancy, prevention of malignancy-related skeletal events
Hypocalcemia, serious infections, skin reactions, osteonecrosis of the jaw
Zoledronate
Zometa†
IV
Inhibits osteoclast function
Hypercalcemia of malignancy, prevention of malignancy-related skeletal events
Kidney damage, osteonecrosis of the jaw
Antiestrogens
Tamoxifen
Use for prevention of breast cancer.
Cytotoxic drugs (chemotherapy)
Denosumab and bisphosphonates for skeletal-related events
Denosumab
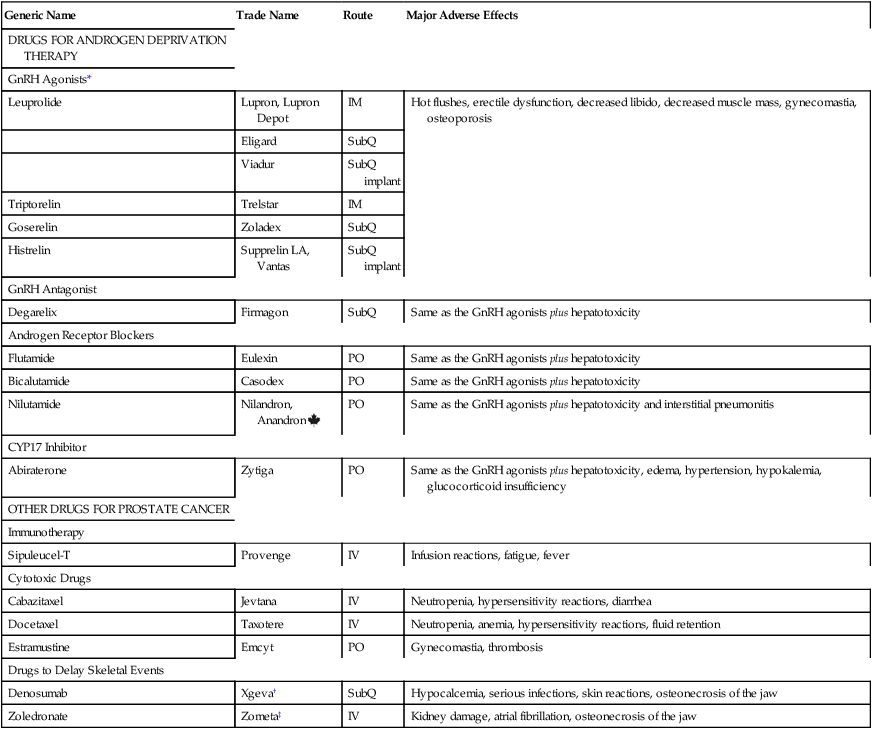
Drugs for prostate cancer

Generic Name
Trade Name
Route
Major Adverse Effects
DRUGS FOR ANDROGEN DEPRIVATION THERAPY
GnRH Agonists*
Leuprolide
Lupron, Lupron Depot
IM
Hot flushes, erectile dysfunction, decreased libido, decreased muscle mass, gynecomastia, osteoporosis
Eligard
SubQ
Viadur
SubQ implant
Triptorelin
Trelstar
IM
Goserelin
Zoladex
SubQ
Histrelin
Supprelin LA, Vantas
SubQ implant
GnRH Antagonist
Degarelix
Firmagon
SubQ
Same as the GnRH agonists plus hepatotoxicity
Androgen Receptor Blockers
Flutamide
Eulexin
PO
Same as the GnRH agonists plus hepatotoxicity
Bicalutamide
Casodex
PO
Same as the GnRH agonists plus hepatotoxicity
Nilutamide
Nilandron, Anandron 
PO
Same as the GnRH agonists plus hepatotoxicity and interstitial pneumonitis
CYP17 Inhibitor
Abiraterone
Zytiga
PO
Same as the GnRH agonists plus hepatotoxicity, edema, hypertension, hypokalemia, glucocorticoid insufficiency
OTHER DRUGS FOR PROSTATE CANCER
Immunotherapy
Sipuleucel-T
Provenge
IV
Infusion reactions, fatigue, fever
Cytotoxic Drugs
Cabazitaxel
Jevtana
IV
Neutropenia, hypersensitivity reactions, diarrhea
Docetaxel
Taxotere
IV
Neutropenia, anemia, hypersensitivity reactions, fluid retention
Estramustine
Emcyt
PO
Gynecomastia, thrombosis
Drugs to Delay Skeletal Events
Denosumab
Xgeva†
SubQ
Hypocalcemia, serious infections, skin reactions, osteonecrosis of the jaw
Zoledronate
Zometa‡
IV
Kidney damage, atrial fibrillation, osteonecrosis of the jaw
Androgen deprivation therapy
Gonadotropin-releasing hormone agonists
Abiraterone, a cyp17 inhibitor
Drug interactions.
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree



Anticancer drugs II: hormonal agents, targeted drugs, and other noncytotoxic anticancer drugs
 ] (an SSRI) and venlafaxine [Effexor] (a serotonin/norepinephrine reuptake inhibitor).
] (an SSRI) and venlafaxine [Effexor] (a serotonin/norepinephrine reuptake inhibitor). ] blocks receptors for androgens. The drug is approved for metastatic prostate cancer in men who have undergone surgical castration. Benefits derive from blocking the actions of adrenal androgens, which are not reduced by castration. In clinical trials, nilutamide reduced bone pain, prolonged progression-free survival, and increased median survival time. The recommended dosage is 300 mg once daily PO for 30 days followed by 150 mg once daily thereafter. Treatment should begin within 24 hours of castration.
] blocks receptors for androgens. The drug is approved for metastatic prostate cancer in men who have undergone surgical castration. Benefits derive from blocking the actions of adrenal androgens, which are not reduced by castration. In clinical trials, nilutamide reduced bone pain, prolonged progression-free survival, and increased median survival time. The recommended dosage is 300 mg once daily PO for 30 days followed by 150 mg once daily thereafter. Treatment should begin within 24 hours of castration.
Get Clinical Tree app for offline access