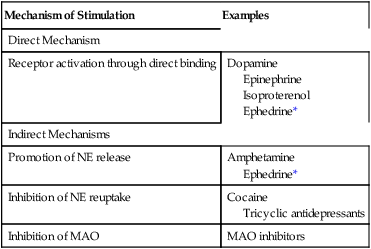
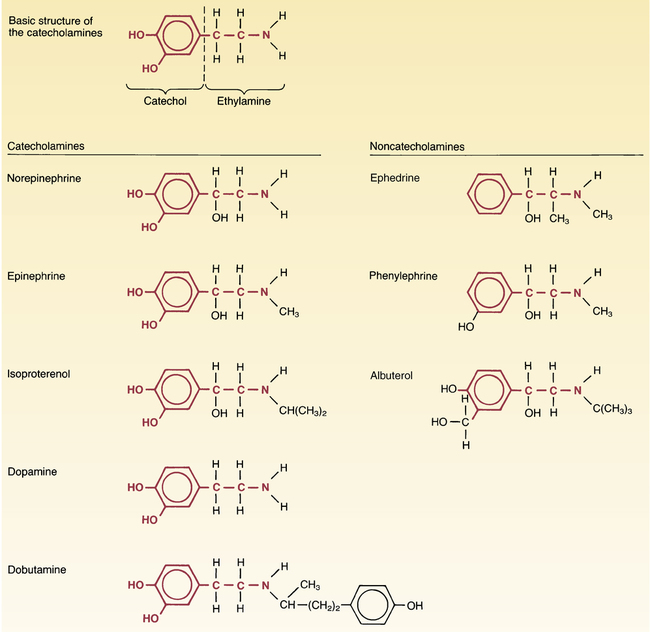
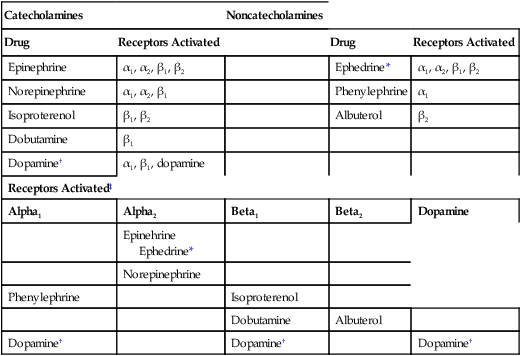
CHAPTER 17 Drugs can activate adrenergic receptors by four basic mechanisms: (1) direct receptor binding, (2) promotion of norepinephrine (NE) release, (3) blockade of NE reuptake, and (4) inhibition of NE inactivation. Note that only the first mechanism is direct. With the other three, receptor activation occurs by an indirect process. Examples of drugs that act by these four mechanisms are presented in Table 17–1. TABLE 17–1 Mechanisms of Adrenergic Receptor Activation MAO = monoamine oxidase, NE = norepinephrine. *Ephedrine is a mixed-acting drug that activates receptors directly and by promoting release of norephinephrine. As discussed in Chapter 13, some of the NE in terminals of adrenergic neurons is subject to inactivation by monoamine oxidase (MAO). Hence, drugs that inhibit MAO can increase the amount of NE available for release, and can thereby enhance receptor activation. (It should be noted that, in addition to being present in sympathetic nerves, MAO is present in the liver and the intestinal wall. The significance of MAO at these other sites is considered later in the chapter.) The catecholamines are so named because they contain a catechol group and an amine group. A catechol group is simply a benzene ring that has hydroxyl groups on two adjacent carbons (Fig. 17–1). The amine component of the catecholamines is ethylamine. Structural formulas for each of the major catecholamines—epinephrine, norepinephrine, isoproterenol, dopamine, and dobutamine—are presented in Figure 17–1. Because of their chemistry, all catecholamines have three properties in common: (1) they cannot be used orally, (2) they have a brief duration of action, and (3) they cannot cross the blood-brain barrier. Catecholamines are polar molecules, and hence cannot cross the blood-brain barrier. (Recall from Chapter 4 that polar compounds penetrate membranes poorly.) The polar nature of the catecholamines is due to the hydroxyl groups on the catechol portion of the molecule. Because they cannot cross the blood-brain barrier, catecholamines have minimal effects on the CNS. The receptor specificities of the major adrenergic agonists are summarized in Table 17–2. In the upper part of the table, receptor specificity is presented in tabular form. In the lower part, the same information is presented schematically. By learning (memorizing) the content of Table 17–2, you will be well on your way toward understanding the pharmacology of the sympathomimetic drugs. TABLE 17–2 Receptor Specificity of Representative Adrenergic Agonists *Ephedrine is a mixed-acting agent that causes NE release and also activates alpha and beta receptors directly. †Receptor activation by dopamine is dose dependent. ‡This chart represents in graphic form the same information on receptor specificity given above. Arrows indicate the range of receptors that the drugs can activate (at usual therapeutic doses). Please note that the concept of receptor specificity is relative, not absolute. The ability of a drug to selectively activate certain receptors to the exclusion of others depends on the dosage: at low doses, selectivity is maximal; as dosage increases, selectivity declines. For example, when albuterol is administered in low to moderate doses, the drug is highly selective for beta2-adrenergic receptors. However, if the dosage is high, albuterol will activate beta1 receptors as well. The information on receptor specificity in Table 17–2 refers to usual therapeutic doses. So-called selective agents will activate additional adrenergic receptors if the dosage is abnormally high. In this section we discuss the responses—both therapeutic and adverse—that can be elicited with sympathomimetic drugs. Since many adrenergic agonists activate more than one type of receptor (see Table 17–2), it could be quite confusing if we were to talk about the effects of the sympathomimetics while employing specific drugs as examples. Consequently, rather than attempting to structure this presentation around representative drugs, we discuss the actions of the adrenergic agonists one receptor at a time. Our discussion begins with alpha1 receptors, and then moves to alpha2 receptors, beta1 receptors, beta2 receptors, and finally dopamine receptors. For each receptor type, we discuss both the therapeutic and adverse responses that can result from receptor activation. To understand the effects of any specific adrenergic agonist, all you need is two types of information: (1) the identity of the receptors at which the drug acts and (2) the effects produced by activating those receptors. Combining these two types of information will reveal a profile of drug action. This is the same approach to understanding neuropharmacologic agents that we discussed in Chapter 12. Before you go deeper into this chapter, I encourage you (strongly advise you) to review Table 13–3. Since we are about to discuss the clinical consequences of adrenergic receptor activation, and since Table 13–3 summarizes the responses to activation of those receptors, the benefits of being familiar with Table 13–3 are obvious. If you choose not to memorize Table 13–3 now, at least be prepared to refer back to it as we discuss the consequences of receptor activation. In this section we discuss the therapeutic and adverse effects that can result from activation of alpha1-adrenergic receptors. As shown in Table 17–2, drugs capable of activating alpha1 receptors include epinephrine, NE, phenylephrine, ephedrine, and dopamine. As discussed in Chapter 13, alpha2 receptors in the periphery are located presynaptically, and their activation inhibits NE release. Several adrenergic agonists (eg, epinephrine, NE) are capable of causing alpha2 activation. However, their ability to activate alpha2 receptors in the periphery has little clinical significance. There are no therapeutic applications related to activation of peripheral alpha2 receptors. Furthermore, activation of these receptors rarely causes significant adverse effects. In contrast to alpha2 receptors in the periphery, alpha2 receptors in the CNS are of great clinical significance. By activating central alpha2 receptors, we can produce two useful effects: (1) reduction of sympathetic outflow to the heart and blood vessels and (2) relief of severe pain. The central alpha2 agonists used for effects on the heart and blood vessels, and the agents used to relieve pain, are discussed in Chapters 19 and 28, respectively. All of the clinically relevant responses to activation of beta1 receptors result from activating beta1 receptors in the heart; activation of renal beta1 receptors is not associated with either beneficial or adverse effects. As indicated in Table 17–2, beta1 receptors can be activated by epinephrine, NE, isoproterenol, dopamine, dobutamine, and ephedrine.
Adrenergic agonists
Mechanisms of adrenergic receptor activation

Mechanism of Stimulation
Examples
Direct Mechanism
Receptor activation through direct binding
Dopamine
Epinephrine
Isoproterenol
Ephedrine*
Indirect Mechanisms
Promotion of NE release
Amphetamine
Ephedrine*
Inhibition of NE reuptake
Cocaine
Tricyclic antidepressants
Inhibition of MAO
MAO inhibitors
Inhibition of ne inactivation.
Overview of the adrenergic agonists
Chemical classification: catecholamines versus noncatecholamines
Catecholamines
 Structures of representative catecholamines and noncatecholamines.
Structures of representative catecholamines and noncatecholamines.
Catecholamines: Note that all of the catecholamines share the same basic chemical formula. Because of their biochemical properties, the catecholamines cannot be used orally, cannot cross the blood-brain barrier, and have short half-lives (owing to rapid inactivation by MAO and COMT).
Noncatecholamines: Although structurally similar to catecholamines, noncatecholamines differ from catecholamines in three important ways: they can be used orally; they can cross the blood-brain barrier; and, because they are not rapidly metabolized by MAO or COMT, they have much longer half-lives.
Receptor specificity

Catecholamines
Noncatecholamines
Drug
Receptors Activated
Drug
Receptors Activated
Epinephrine
α1, α2, β1, β2
Ephedrine*
α1, α2, β1, β2
Norepinephrine
α1, α2, β1
Phenylephrine
α1
Isoproterenol
β1, β2
Albuterol
β2
Dobutamine
β1
Dopamine†
α1, β1, dopamine
Receptors Activated‡
Alpha1
Alpha2
Beta1
Beta2
Dopamine
Epinehrine
Ephedrine*
Norepinephrine
Phenylephrine
Isoproterenol
Dobutamine
Albuterol
Dopamine†
Dopamine†
Dopamine†
Therapeutic applications and adverse effects of adrenergic receptor activation
Clinical consequences of alpha1 activation
Clinical consequences of alpha2 activation
Clinical consequences of beta1 activation
Therapeutic applications of beta1 activation
< div class='tao-gold-member'>
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Adrenergic agonists
Only gold members can continue reading. Log In or Register to continue
Get Clinical Tree app for offline access
