Care of Patients with Hematologic Disorders
Objectives
1. Identify the causes of the various types of anemias.
2. Develop a plan of care for the patient with an anemia.
3. Explain the pathophysiology and care of sickle cell disease.
4. Compare cell abnormalities of polycythemia vera to those of leukemia.
5. Formulate a teaching plan for the patient with leukemia.
6. Comprehend why multiple myeloma is a disease affecting older people.
7. Discuss the problems and treatments the hemophilia patient faces.
3. Perform an assessment on a patient with a suspected hematologic disorder.
4. Assist with the development of a plan of care for an adult with leukemia.
5. Assess for signs and symptoms of disseminated intravascular coagulation.
Key Terms
allogeneic (ĂL-ō-JĔN-ĭk, p. 364)
anemia (ă-NĒ-mē-ă, p. 346)
autologous (ăw-TŎL-ŏ-gŭs, pp. 362, 364)
disseminated intravascular coagulation (DIC) (dĭ-SĔM-ĭ-nāt-ĕd ĭn-tră-VĂS-cū-lăr kō-ăg-ū-LĀ-shŭn, p. 362)
ecchymoses (ĕk-ĭ-MŌ-sēz, p. 359)
hemarthrosis (hē-măr-THRŌ-sĭs, p. 361)
hemolysis (hē-MŎL-ĭ-sĭs, p. 347)
hypovolemia (hī-pō-vō-LĒ-mē-ă, p. 346)
leukapheresis (lū-kă-fĕ-RĒ-sĭs, p. 356)
purpura (PŬR-pū-ră, p. 359)
splenomegaly (splē-nō-MĒG-ă-lē, p. 354)
stomatitis (stō-mă-TĪ-tĭs, p. 359)
thrombocytopenia (thrŏm-bō-sīt-ō-PĒ-nē-ă, p. 359)
 http://evolve.elsevier.com/deWit/medsurg
http://evolve.elsevier.com/deWit/medsurg
Disorders of the Hematologic System
Anemia
In the human body, healthy red blood cells (RBCs) carry oxygen to tissues. A balance is maintained between the production of new RBCs and the disposal of old “worn-out” RBCs. Anemia occurs when something interferes with this balance or interferes with the maturation of cells. Anemia is a state in which there are insufficient numbers of functioning RBCs, or a lack of hemoglobin, to meet the demands of the tissues for oxygen.
Etiology
There are three major classifications of anemia, according to cause:
• Anemia resulting from blood loss
• Anemia resulting from a failure in blood cell production
• Anemia associated with an excessive destruction of red cells
Rapid, severe bleeding leads to anemia from blood loss, hypovolemia (decreased volume of circulating blood), and, potentially, shock. A blood loss that leads to anemia may result from severe trauma to the blood vessels and massive hemorrhage or the blood loss may be more gradual, as from a small, bleeding peptic ulcer that causes a chronic blood loss.
The amount of blood loss that leads to hypovolemic shock varies, depending on the ability of the patient’s body to compensate for the lost fluid volume. A blood loss of even 500 mL in an adult who had normal circulating volume may cause hypovolemic shock. See Chapter 45 for the treatment of shock. Table 17-1 shows the amount of blood loss and consequent clinical manifestations.
Table 17-1
Clinical Manifestations of Acute Blood Loss
| Volume Lost | Clinical Manifestations |
| 10% | None |
| 20% | At rest, no signs or symptoms; slight postural hypotension when standing; tachycardia with exercise |
| 30% | Blood pressure and pulse normal when supine; postural hypotension and tachycardia with exercise |
| 40% | Below-normal blood pressure, central venous pressure, and cardiac output at rest; rapid, thready pulse and cold, clammy skin |
| 50% | Shock and potential death |
Adapted from Lewis, S.L., Heitkemper, M.M., Dirksen, S.R., et al. (2007). Medical-Surgical Nursing: Assessment and Management of Clinical Problems (7th ed.). St. Louis: Mosby, p. 695.
Anemia caused by a failure in cell production is the result of either a deficiency of certain substances necessary for the formation of RBCs, or results from the abnormal function of bone marrow. Examples of this type of anemia are:
• Anemia resulting from bone marrow suppression caused by toxic substances
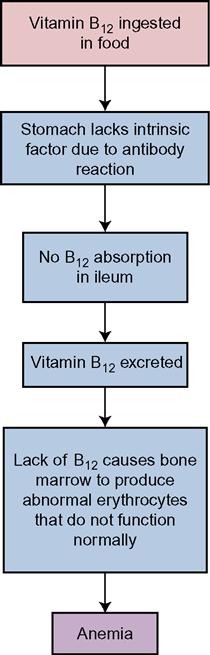
• Pernicious anemia, in which there is faulty absorption of specific nutrients, such as vitamin B12
Iron or folic acid may not be well absorbed in people who have an intestinal malabsorption syndrome.
Hemolytic anemias, in which red cells are destroyed prematurely in the body, have many causes. Hemolytic anemia can be a result of genetic defects that affect cell structure, causing the cells to disintegrate quickly. Some of the hemolytic anemias, such as thalassemia, are inherited, whereas others are acquired when erythrocytes are exposed to poisonous agents, such as chemicals or certain bacterial toxins.
Immune reactions can cause blood cell hemolysis (destruction of red cells). The presence of toxins in the blood, infections such as malaria, transfusion reactions, and changes in blood chemistry may cause red cell hemolysis. Blood incompatibility in the newborn (erythroblastosis fetalis) is another cause.

There is about a 20% incidence of anemia among the elderly, most often because of poor nutrition. Shock may develop with smaller blood loss in this group because of decreased vascular tone and impaired cardiac function.
Pathophysiology
Iron deficiency anemia occurs when total body iron is insufficient and erythropoiesis is diminished. The lack of iron impedes the formation of hemoglobin (Hb) (Concept Map 17-1). In pernicious anemia, an autoimmune disease, the intrinsic factor is missing from the gastric juices, and vitamin B12 is not absorbed without it. Vitamin B12 acts as a coenzyme in conjunction with folate metabolism and is important in the utilization of iron and protein for the manufacture of RBCs. The result of the missing intrinsic factor is that the red cell production is decreased, and those red cells that are produced are abnormal in their structure and function (Concept Map 17-2). To correct this condition, the physician will order the administration of vitamin B12. A folic acid deficiency also contributes to anemia (Gentili et al., 2009).
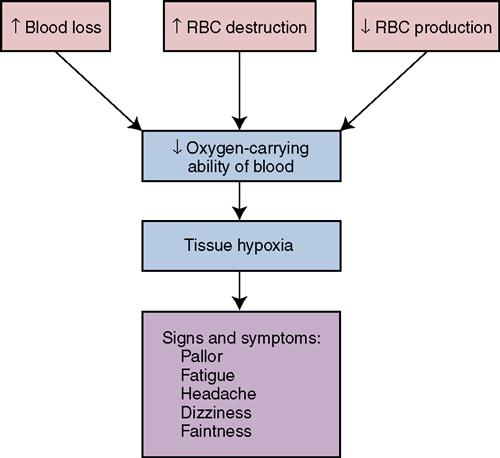
Hemolytic anemias associated with excessive destruction of RBCs are quite rare. When red cells are not normal, they break up easily or are destroyed by the body more quickly than are normal red cells. This RBC destruction causes the anemia.
Anemia occurs in end-stage renal disease patients when there is a deficiency of production of erythropoietin, a substance necessary to stimulate the production of RBCs in the bone marrow. This problem is usually corrected by the administration of epoetin alfa (Epogen), which stimulates red cell production (Singh, 2006). Oxygen transport depends on the number and condition of the red cells and the amount of hemoglobin they contain.

Signs and Symptoms
Signs and symptoms of anemias from causes other than rapid bleeding depend on whether the anemia is mild, moderate, or severe. Signs and symptoms of mild anemia (Hb 9.5 to 13 g/dL) are mild headache, palpitations, and dyspnea on exertion. Moderate anemia (Hb 6 to 10 g/dL) may include brittle nails, sore tongue, pallor, chronic fatigue, headache, and dizziness or faintness. Table 17-2 presents the many signs and symptoms of severe anemia. Tachypnea and tachycardia may develop with severe anemia due to the decreased ability of the blood to transport sufficient oxygen to the tissues.
Table 17-2
Signs and Symptoms of Severe Anemia
| Body System | Signs and Symptoms |
| General | Sensitivity to cold, lethargy, weight loss |
| Eyes | Blurred vision, blue colored sclera, yellowing of conjunctiva or pale conjunctiva, retinal hemorrhage |
| Skin | Pallor of face and palms, pruritus, jaundice, pale nail beds, pale mucous membranes, stomatitis, brittle nails, cheilitis |
| Cardiovascular | Palpitations, tachycardia, angina, systolic murmur, widened pulse pressure, intermittent claudication, CHF, possible MI |
| Respiratory | Tachypnea, orthopnea, dyspnea at rest |
| Gastrointestinal | Anorexia, difficulty swallowing, glossitis, enlarged liver, enlarged spleen, smooth tongue |
| Musculoskeletal | Bone pain |
| Neurologic | Headache, dizziness, impaired thinking, irritability, depression, fatigue |
Diagnosis
The microscopic appearance of the red cells in a film of blood that has been spread over a slide (a peripheral smear) gives information about abnormalities in size, shape, and color of erythrocytes circulating in the patient’s bloodstream. The complete blood count (CBC) and differential cell count results are used to diagnose the presence of anemia. Measuring the quantity of hemoglobin tells whether the cells have sufficient amounts of hemoglobin to carry adequate oxygen to the body.
The prefix normo- refers to normal; the suffix -cyte refers to cells; the suffix -chrom refers to color; and the suffix –ic means “having the quality of” or “characterized by.” Thus a normocytic, normochromic anemia is characterized by cells that are normal in size and color, but that have a deficiency in the number of RBCs and a low hematocrit. This type of anemia usually occurs as a result of sudden blood loss.
A hypochromic, microcytic anemia is characterized by decreased levels of hemoglobin (not enough color) and small (micro) cells. This type of anemia is typical of an iron deficiency anemia.

Common Foods High in Iron and Folic Acid
Foods High in Iron
Adding raw spinach to dinner salads and snacking on raisins or dried apricots can quickly improve iron intake. Iron-enriched cereals and breads also can be added to the diet.
Foods High in Folic Acid
Note: Many of the foods high in iron also are high in folic acid.
Treatment
Anemia from chronic, slow blood loss is treated by correcting the underlying problem and then building replacement blood cells. Anemia caused by inadequate iron, folic acid, or protein intake is managed with oral iron supplements, vitamins, and diet adjustment. If the anemia is serious, blood transfusions may be given, or iron supplementation may be administered intravenously (IV) with iron dextran (Imferon), sodium ferrous gluconate, or iron sucrose.
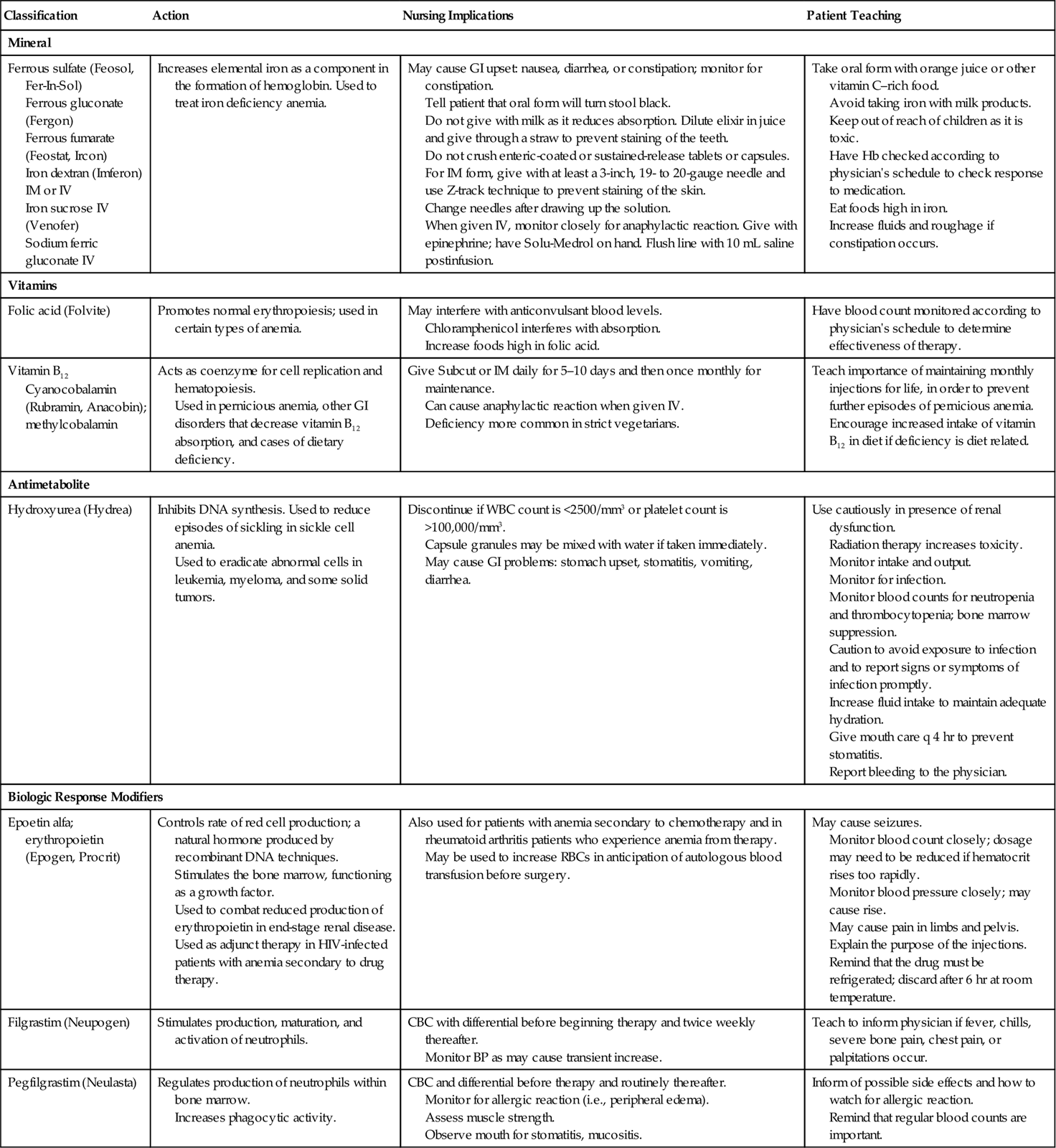
Pernicious anemia is treated by regular injections of vitamin B12, or by weekly use of an intranasal form of cyanocobalamin (Nascobal), as the deficiency of intrinsic factor prevents adequate absorption of this vitamin from food. There should be sufficient folic acid in the diet or by supplement. Table 17-3 presents the medications most commonly prescribed for hematologic disorders.
 Table 17-3
Table 17-3
Medications Commonly Prescribed for Disorders of the Hematologic System*
| Classification | Action | Nursing Implications | Patient Teaching |
| Mineral | |||
| Ferrous sulfate (Feosol, Fer-In-Sol) Ferrous gluconate (Fergon) Ferrous fumarate (Feostat, Ircon) Iron dextran (Imferon) IM or IV Iron sucrose IV (Venofer) Sodium ferric gluconate IV | Increases elemental iron as a component in the formation of hemoglobin. Used to treat iron deficiency anemia. | May cause GI upset: nausea, diarrhea, or constipation; monitor for constipation. Tell patient that oral form will turn stool black. Do not give with milk as it reduces absorption. Dilute elixir in juice and give through a straw to prevent staining of the teeth. Do not crush enteric-coated or sustained-release tablets or capsules. For IM form, give with at least a 3-inch, 19- to 20-gauge needle and use Z-track technique to prevent staining of the skin. Change needles after drawing up the solution. When given IV, monitor closely for anaphylactic reaction. Give with epinephrine; have Solu-Medrol on hand. Flush line with 10 mL saline postinfusion. | Take oral form with orange juice or other vitamin C–rich food. Avoid taking iron with milk products. Keep out of reach of children as it is toxic. Have Hb checked according to physician’s schedule to check response to medication. Eat foods high in iron. Increase fluids and roughage if constipation occurs. |
| Vitamins | |||
| Folic acid (Folvite) | Promotes normal erythropoiesis; used in certain types of anemia. | May interfere with anticonvulsant blood levels. Chloramphenicol interferes with absorption. Increase foods high in folic acid. | Have blood count monitored according to physician’s schedule to determine effectiveness of therapy. |
| Vitamin B12 Cyanocobalamin (Rubramin, Anacobin); methylcobalamin | Acts as coenzyme for cell replication and hematopoiesis. Used in pernicious anemia, other GI disorders that decrease vitamin B12 absorption, and cases of dietary deficiency. | Give Subcut or IM daily for 5–10 days and then once monthly for maintenance. Can cause anaphylactic reaction when given IV. Deficiency more common in strict vegetarians. | Teach importance of maintaining monthly injections for life, in order to prevent further episodes of pernicious anemia. Encourage increased intake of vitamin B12 in diet if deficiency is diet related. |
| Antimetabolite | |||
| Hydroxyurea (Hydrea) | Inhibits DNA synthesis. Used to reduce episodes of sickling in sickle cell anemia. Used to eradicate abnormal cells in leukemia, myeloma, and some solid tumors. | Discontinue if WBC count is <2500/mm3 or platelet count is >100,000/mm3. Capsule granules may be mixed with water if taken immediately. May cause GI problems: stomach upset, stomatitis, vomiting, diarrhea. | Use cautiously in presence of renal dysfunction. Radiation therapy increases toxicity. Monitor intake and output. Monitor for infection. Monitor blood counts for neutropenia and thrombocytopenia; bone marrow suppression. Caution to avoid exposure to infection and to report signs or symptoms of infection promptly. Increase fluid intake to maintain adequate hydration. Give mouth care q 4 hr to prevent stomatitis. Report bleeding to the physician. |
| Biologic Response Modifiers | |||
| Epoetin alfa; erythropoietin (Epogen, Procrit) | Controls rate of red cell production; a natural hormone produced by recombinant DNA techniques. Stimulates the bone marrow, functioning as a growth factor. Used to combat reduced production of erythropoietin in end-stage renal disease. Used as adjunct therapy in HIV-infected patients with anemia secondary to drug therapy. | Also used for patients with anemia secondary to chemotherapy and in rheumatoid arthritis patients who experience anemia from therapy. May be used to increase RBCs in anticipation of autologous blood transfusion before surgery. | May cause seizures. Monitor blood count closely; dosage may need to be reduced if hematocrit rises too rapidly. Monitor blood pressure closely; may cause rise. May cause pain in limbs and pelvis. Explain the purpose of the injections. Remind that the drug must be refrigerated; discard after 6 hr at room temperature. |
| Filgrastim (Neupogen) | Stimulates production, maturation, and activation of neutrophils. | CBC with differential before beginning therapy and twice weekly thereafter. Monitor BP as may cause transient increase. | Teach to inform physician if fever, chills, severe bone pain, chest pain, or palpitations occur. |
| Pegfilgrastim (Neulasta) | Regulates production of neutrophils within bone marrow. Increases phagocytic activity. | CBC and differential before therapy and routinely thereafter. Monitor for allergic reaction (i.e., peripheral edema). Assess muscle strength. Observe mouth for stomatitis, mucositis. | Inform of possible side effects and how to watch for allergic reaction. Remind that regular blood counts are important. |
For hemolytic anemia, the underlying cause is found and corrected (if possible) and then the blood volume is rebuilt with added iron and appropriate diet. If the anemia is severe, blood transfusion may be indicated.

Your patient who has suffered a blood loss and is now anemic complains that he is short of breath. Can you explain how blood loss might affect respiration?
Nursing Management
Assessment (Data Collection)
Whenever a patient complains of fatigue, headaches, or shortness of breath, anemia should always be considered. Besides the CBC results, data regarding physical signs and symptoms are collected.

Data Collection When Anemia Is Suspected
Health History
Ask the patient the following questions:
• Have you had any recent blood loss or trauma?
• Do you have chronic liver, endocrine, gastrointestinal, or renal disease?
• What medications, vitamins, supplements, or herbal products do you take?
• What surgeries have you had and when?
• Have you ever had radiation treatments or chemotherapy?
• Is there a history of genetic blood disorders in your family?
• Has your appetite or weight changed?
• Have you noticed any changes in your urine or stool?
• Are you experiencing shortness of breath, weakness, or fatigue?
• Have you noticed any heart palpitations?
• Do you get frequent headaches?
• Have you noticed any changes in vision or dizziness?
• Do you have pain or itching anywhere?
Physical Assessment
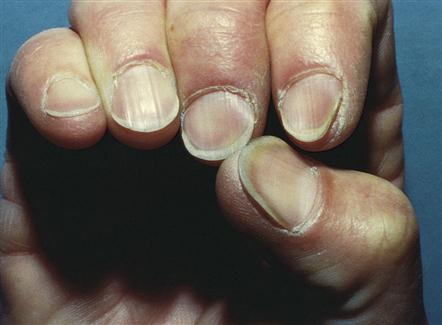
• Skin: Pale skin and mucous membranes; pale conjunctiva, yellowing of sclera; cracks in lips; brittle, spoon-shaped fingernails (Figure 17-1); jaundice; petechiae; ecchymoses; dry, brittle, thinning hair
• Respiratory: Tachypnea, orthopnea, dyspnea on exertion or at rest
• Cardiac: Tachycardia, systolic murmur, angina, ankle edema
• Neurologic: Headache, dizziness, confusion, irritability, ataxia (unsteady gait), paresthesia
Pertinent Laboratory Values
Nursing Diagnosis
Nursing diagnoses are chosen based on the clinical findings and problems identified. Common nursing diagnoses include:
Planning
Expected outcomes are written for the specific individual nursing diagnoses chosen to resolve the patient’s problems. For the nursing diagnoses listed above, outcomes might include:
Implementation
Intervention is based on an understanding of the particular kind of anemia affecting the patient. Anemia from blood loss presents problems quite different from those related to chronic—and possibly incurable—aplastic or hemolytic anemia. For patients with anemias that interfere with clotting and that tend to cause bleeding episodes, nursing actions are directed toward preventing the episodes. For any patient with anemia severe enough to cause fatigue, assist with daily living activities, and provide planned rest periods.
Nursing functions include administering blood, iron, vitamin B12, and folic acid and monitoring for desired effects. Patients are educated about needed dietary adjustments. Patients should be taught that iron is absorbed more readily if vitamin C is simultaneously present in the gastrointestinal (GI) system. Taking iron medication with orange juice provides the necessary vitamin C.
Analgesia for headache or joint pain is given as ordered, and the patient is monitored for adverse side effects. More nursing diagnoses commonly associated with hematologic problems, including anemia, and lists of appropriate interventions are included in Table 16-2.

Evaluation
Evaluation data are gathered to determine whether expected outcomes are being met. Laboratory values are particularly important when evaluating the care of the patient with anemia. However, equally important are data showing that the problems caused by the anemia are resolving.
Aplastic Anemia
Aplastic anemia (a rare disorder) may develop after a viral infection, as a reaction to a drug, or because of an inherited tendency. The disease is characterized by bone marrow depression and is thought to probably be an immune-mediated disease. Red cells, white cells, and platelet levels are decreased. The toxic effects of certain substances can be responsible for aplastic anemia. Some of these agents include benzene; insecticides; drugs, such as chloramphenicol (Chloromycetin), phenylbutazone (Butazolidin), and sulfonamides; some anticonvulsants; gold compounds used to treat rheumatoid arthritis; and alkylating agents or antimetabolites used in chemotherapy. Many other drugs  can cause aplastic anemia, but this adverse effect is rare. Radiation exposure is another factor in the development of the disorder.
can cause aplastic anemia, but this adverse effect is rare. Radiation exposure is another factor in the development of the disorder.

Monitor Drug Side Effects
It is your responsibility to monitor blood studies carefully for all patients who are receiving any drug that is potentially damaging to the bone marrow.

What chemical products in your home or garage are capable of causing bone marrow depression?
Impairment or failure of bone marrow function leading to the loss of stem cells is the cause of aplastic anemia (Concept Map 17-3). With aplastic anemia, the bone marrow has decreased cells and increased fatty tissue. In addition to the signs and symptoms of iron deficiency anemia, ecchymosis, petechiae, and hemorrhage related to low platelet count also occur. Infection is frequent and may not cause an inflammatory response because of the very low leukocyte count. There is often frequent bleeding in the mouth.
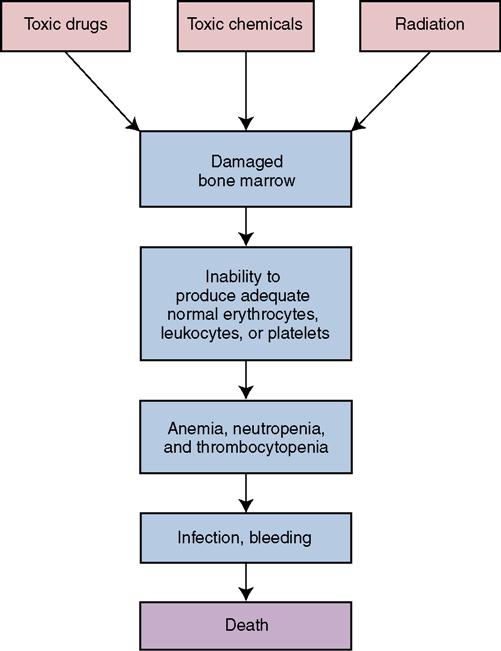
Diagnosis is by blood count with differential, bone marrow biopsy, and ruling out other disorders. Aplastic anemia causes an emergency situation. Treatment must eliminate any identifiable underlying cause. Packed red cells and platelets are administered. Antibiotics are given for identified infection; oxygen is sometimes administered to patients with low erythrocyte counts. Bone marrow transplantation (BMT) is the treatment of choice for those under 45 years of age with severe bone marrow depression, but there must be an identical human leukocyte antigen (HLA) match. Immunosuppressive therapy with antithymocyte globulin (ATG) and cyclosporine is showing promise at improving outcomes. ATG contains polyclonal antibodies against human T cells.
Prevention of hemorrhage and infection is a top priority. Psychological support of the patient and family is important when they are faced with this life-threatening condition. Safety measures are priorities. Actions for problems of weakness and fatigue are the same as those presented for anemia earlier in the chapter. Other common nursing interventions are included in Table 16-2. See Chapter 8 and the Evolve website for precautions and actions for the patient with leukopenia and neutropenia and for safety measures when thrombocytopenia is  present.
present.

Dangers of Toxic Agents
All nurses should promote public education about the dangers of toxic agents. It is vitally important that people read and follow the label instructions on all cleaning agents, insecticides, and chemical compounds.
Sickle Cell Disease
Etiology
Sickle cell disease is a genetic disorder in which the gene is inherited from both parents (homozygous gene) (Table 17-4). Sickle cell disease is characterized by erythrocytes that contain more hemoglobin S than hemoglobin A. Sickle cell disease is found in less than 1% of African American newborns, but also affects some people whose ancestors are from the Mediterranean region, the Middle East, and India. Approximately 8% of African Americans carry the gene.
from both parents (homozygous gene) (Table 17-4). Sickle cell disease is characterized by erythrocytes that contain more hemoglobin S than hemoglobin A. Sickle cell disease is found in less than 1% of African American newborns, but also affects some people whose ancestors are from the Mediterranean region, the Middle East, and India. Approximately 8% of African Americans carry the gene.
Table 17-4
Comparison of Four Types of Anemia
| Anemia | Characteristic RBC | Etiology | Additional Effects |
| Iron deficiency anemia | Microcytic, hypochromic Decreased hemoglobin production | Decreased dietary intake, malabsorption, blood loss | Only effects of anemia |
| Pernicious anemia | Megaloblasts, immature nucleated cells | Deficit of intrinsic factor due to immune reaction | Neurologic damage Achlorhydria |
| Aplastic anemia | Often normal cells Pancytopenia | Bone marrow damage or failure | Excessive bleeding and multiple infections |
| Sickle cell anemia | RBC elongates and hardens in “sickle” shape when O2 levels are low—short life span | Recessive inheritance | Painful crises with multiple infarctions Hyperbilirubinemia |
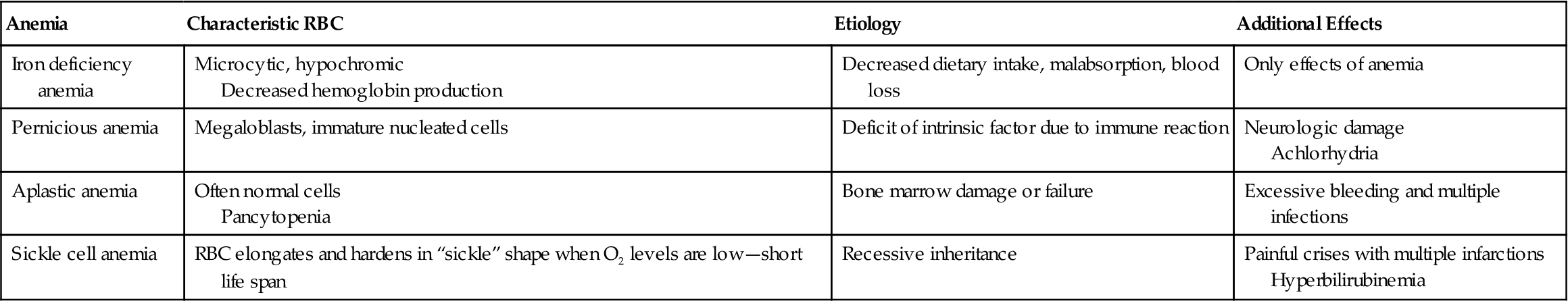
From Gould, B.E. & Dyer, R. M. (2011). Pathophysiology for the Health Professions (4th ed., p. 259). Philadelphia: Saunders.
Sickle cell trait, in which only about 50% of an individual’s total hemoglobin is affected, is present in about 10% of the African American population of the United States. The trait is heterozygous, meaning that the person has an inherited gene for the trait from one parent only. People with the heterozygous trait for sickle cell are carriers; they can transmit the gene to their children even when they themselves do not show signs of the disease. Therefore, genetic counseling and adequate screening for early detection of the disease are considered extremely important to control sickle cell anemia. In the United States, many patients with sickle cell anemia live into their mid-40s. The most common cause of death is acute chest syndrome, where damage occurs to the lungs.
Pathophysiology
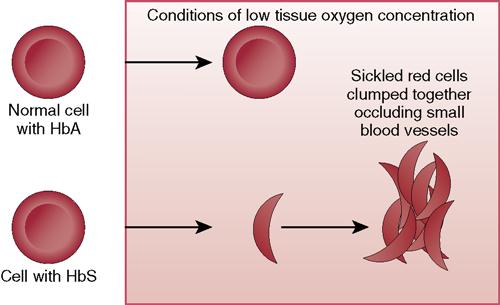
When the patient with sickle cell disease experiences lower oxygenation than normal, the defective S hemoglobin forms clumps in the red cells, causing them to assume a sickle shape, blocking blood vessels, breaking apart, and forming thrombi that cause organ damage. Sickle cells are destroyed by the body very quickly, causing anemia.
Sickle cell trait occurs in people who have only one gene, rather than a pair of genes, for sickle cell anemia. They usually do not have problems with cells assuming a sickle shape unless they experience severe oxygen deficiency.
Signs and Symptoms
The signs and symptoms of sickle cell disease are those that indicate lack of oxygen and blood flow, such as pallor, lethargy, and pain. The problems from interrupted normal blood flow affect many organs (Figure 17-2). Painful swelling of the hands and feet related to bone infarction from the sickled cells (hand-foot syndrome) may occur. After sickle cell crisis, signs typical of anemia occur because the abnormally shaped cells are very fragile, break easily, and are destroyed. The RBC and hemoglobin counts can drop very quickly during a crisis.
Diagnosis
A peripheral blood smear can show sickled cells. The sickling test, which exposes RBCs to a deoxygenating agent, is diagnostic. Hemoglobin electrophoresis identifies the presence of abnormal hemoglobin. During crisis, there will be elevations of serum bilirubin because of the hemolysis of the abnormal red cells. Bone and joint abnormalities are revealed by skeletal x-rays.
Treatment
There is no cure or specific treatment for sickle cell anemia; treatment is primarily symptomatic and preventive. Patients should be taking folic acid regularly and eating a diet with sufficient protein to help build red cells. Infection is to be avoided, and the patient should receive all recommended immunizations against influenza, hepatitis A and B, pneumonia, tetanus, and the like. Adequate intake of fluid on a daily basis is important to keep the blood as fluid as possible. Alcohol and recreational drugs are to be avoided as they can cause complications. Quick attention for illness should be sought.
The drug hydroxyurea (Hydrea) has been found to reduce the frequency of sickling episodes. Patients on this drug have shown a 50% decrease in the number of hospitalizations for crisis (Platt, 2008). Sodium cromoglycate given by inhalation or via nasal route has been shown to significantly reduce the percentage of cells that sickle in venous blood (Bizumukama et al., 2009). If a crisis occurs, the patient may be treated at home with bed rest, adequate fluid intake, and analgesics. Pain control is important during a crisis. Narcotic analgesia with morphine is administered on a continuous basis, usually by patient-controlled analgesia pump. If the patient’s hemoglobin drops considerably or his condition suddenly deteriorates, he is hospitalized, given oxygen, and transfused with packed red cells; in addition, IV fluids are given. An attempt is made to mobilize the sickled cells and to prevent damage to major organs. Infection is treated with appropriate antibiotics.
There are many complications of sickle cell disease, including cholecystitis, stroke, congestive heart failure, and damage to all major organs (Figure 17-3). One of the most common problems is leg ulcers, from impaired circulation to the legs and feet. Protecting the feet and lower legs from injury is important, since small wounds tend to develop into difficult-to-heal ulcers.
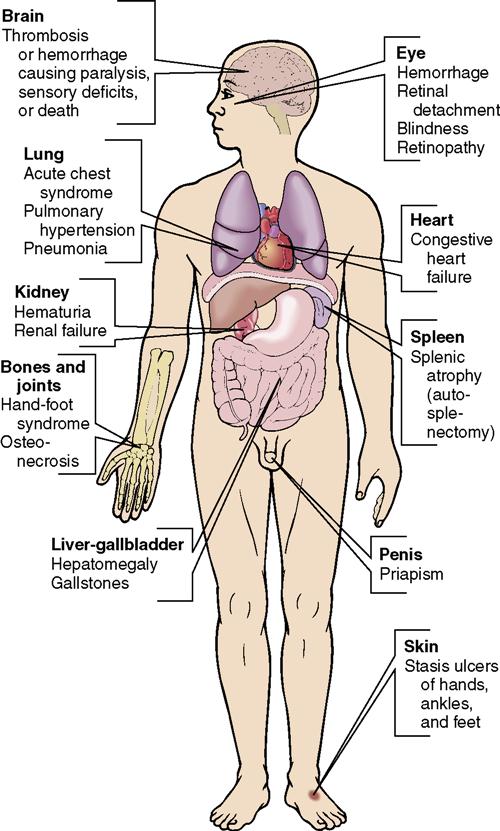
BMT is the only available treatment that can cure some patients. The scarcity of donors, the cost, and the risks involved greatly limit the use of this option. Gene therapy is offering hope for future treatment of sickle cell disease.
Nursing Management
Nursing care is aimed at relieving the symptoms from complications of the disease and minimizing organ damage. Patients are taught to avoid high altitudes, vigorous exercise, and iced liquids. Patients are to maintain adequate fluid intake, refrain from smoking, and obtain treatment for infections promptly. Adequate rest is important as patients with sickle cell anemia experience fatigue. Assessment for adequate pain relief is a top priority  (Arnold & Besa, 2010). Intake and output will be monitored to prevent overloading the patient with fluid. Oxygen therapy is instituted if the patient is hypoxic (oxygen therapy helps prevent further cellular
(Arnold & Besa, 2010). Intake and output will be monitored to prevent overloading the patient with fluid. Oxygen therapy is instituted if the patient is hypoxic (oxygen therapy helps prevent further cellular damage).
damage).
Polycythemia Vera
Excessive production of RBCs results in polycythemia vera. White cell numbers also increase, but not to the degree that they do in leukemia. The cause of polycythemia vera is unknown, but the disease is considered a neoplastic disorder. The blood becomes thick from the increased numbers of cells, blood vessels become distended, and blood flow is sluggish. Because of the sluggish flow, there is a tendency to develop blood clots. Blood pressure is elevated and the heart hypertrophies. Hemorrhage is frequent in areas of distended blood vessels. Signs and symptoms of polycythemia vera include a reddish face with deep-red purplish lips, fatigue, weakness, dizziness, headache, enlarged spleen (splenomegaly), and congested liver. Minor injury may result in excessive bleeding.
Treatment is aimed at reducing the number of blood cells. Phlebotomy, antineoplastic agents, and radiation therapy are all used. In phlebotomy, a blood vessel is pierced, and blood is drawn off. As much as 500 mL of blood at a time may be withdrawn every 2 to 3 months. Increased fluid intake is essential to decrease blood viscosity, and aspirin is used to decrease platelet clumping and clot formation.
A secondary polycythemia may develop in response to prolonged hypoxia and increased erythropoietin secretion. Secondary polycythemia does not have the same effects as primary polycythemia.
Leukemia
The word leukemia, translated literally, means “white blood.” Actually, the white blood cells (WBCs) would have to number 1,000,000/mm3 before the blood would have a milky white appearance, and, although leukemia is characterized by an increase in the number of leukocytes, their number rarely rises above 500,000/mm3. In addition to the increase in number, however, the leukocytes of the patient with leukemia are abnormal cells that do not function as normal white cells do.
Etiology
Leukemia is a cancer, and as with other types of cancers, the exact cause of leukemia is not known. There are factors considered to be closely linked with the development of leukemia. Exposure to ionizing radiation in relatively large doses is one such factor. Another is exposure to certain chemicals, such as benzene, that are toxic to bone marrow. Benzene is an ingredient in lead-free gasoline, and the incidence of leukemia has risen since lead-free gasoline has been in use. The amount of exposure to benzene and other chemicals that causes bone marrow suppression is unknown, and this amount possibly varies among individuals. The point is to be careful about breathing gasoline fumes and using household chemicals and pesticides. The third factor is the retrovirus known as human T-lymphotropic virus 1 (HTLV-1), which causes human T-cell leukemia. People with an abnormal number of chromosomes and chromosomal translocations are at a greater risk for developing acute lymphocytic leukemia. About 90% of patients with chronic myelogenous leukemia have the Philadelphia chromosome.
Malignant production of WBCs is the actual cause of the disease. DNA become damaged. Table 17-5 shows the clinical manifestations of leukemia and the factors linked to their development.
Table 17-5
Causes of Clinical Signs of Leukemia
| Manifestations | Causes |
| Severe infections | Immature and abnormally functioning leukocytes, even though there is an increased number of them. |
| Symptoms of anemia | Rapidly proliferating white cells apparently “crowd out” developing red cells and platelets. |
| Enlarged spleen, liver, and lymph nodes | Excess white cells accumulate within organs, causing distention of tissues. |
| Weakness, pallor, and weight loss due to elevated metabolic rate | Increased production of white cells requires large amounts of amino acids and vitamins. Increased destruction of cells leads to more metabolic wastes that must be disposed of by the body. |
| Renal pain, urinary stones and obstruction to flow of urine, and urinary tract infection | Large amounts of uric acid are released when white cells are destroyed by antileukemic drugs. |
| Headache, disorientation, and other central nervous system symptoms | Abnormal white cells infiltrate the central nervous system. |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


