Chapter 5 Viral and host determinants of HIV-1 disease progression
Specific (Adaptive) Antiviral Immune Response
Humoral immune response
Upon infection with HIV-1, both cellular and humoral immune responses are mounted against the virus. The humoral immune response initially consists mainly of binding antibodies. Only after a few weeks to months, neutralizing antibodies emerge [1, 2]. The importance of neutralizing antibodies in controlling infection was indicated in SIV-infected macaques by B cell depletion [3–5], albeit not in all studies [6]. Moreover, passive transfer of three broadly neutralizing antibodies resulted in a delay in viral rebound after cessation of antiretroviral therapy in acutely HIV-1-infected individuals [7]. However, HIV-1 can escape from neutralizing antibodies and a neutralization-resistant phenotype is associated with changes in the viral envelope [2, 8–10]. The existence of this escape mechanism was first demonstrated in macaques [11] and in a laboratory worker who became accidentally infected with the neutralization-sensitive HIV-1 variant IIIB and in whom a neutralization-resistant IIIB-related variant evolved [12]. A major component of the viral escape mechanism is the increase and repositioning of glycans, the so-called glycan shield [1, 2]. Glycans are strategically positioned and repositioned continuously throughout infection, in a way that minimally affects the interaction of the envelope with the receptor molecules on the cell surface, but maximally hinders the binding of neutralizing antibodies [2]. Additionally, elongation of variable loops in Env has been associated with neutralization resistance [9, 10, 13, 14].
Despite the rapid escape of HIV-1, the neutralizing antibody response was initially still considered to be important because escape from humoral immunity was thought to coincide with a loss of viral fitness [15], which would give rise to a lower viral load set point and hence an ameliorated disease course. However, later studies found that escape from neutralizing antibodies did not coincide with a reduction in replication fitness [16]. In recent years, interest has increased for broadly neutralizing antibodies. These antibodies can neutralize HIV-1 variants from different subtypes and are therefore considered to be directed against conserved epitopes. Based on this it was hypothesized that escape from these broadly neutralizing antibodies would be extremely difficult for the virus and would come at an extreme fitness cost. Surprisingly, escape from autologous serum with broadly neutralizing antibodies occurred rapidly and was also not associated with a loss in viral replication fitness [10]. This is most likely because escape is not associated with mutations in the epitope itself, but rather with the above-mentioned changes in the viral envelope, being an increased number of glycans and an increased length of the variable loops that may occlude the antibody epitope. In agreement with the rapid escape, the presence of even broadly neutralizing activity in serum was not associated with a prolonged asymptomatic course of infection [17, 18].
Cellular immune response: cytotoxic T lymphocytes
HIV-1 specific cellular immune responses, mediated by cytotoxic and helper T lymphocytes, emerge very rapidly after infection [19–21]. Several observations have suggested that HIV-1 specific CD8 cytotoxic T cells (CTLs) contribute to the control of HIV infection. Strong CTL responses are often observed in long-term asymptomatic HIV-1-infected individuals but diminish with progression to AIDS [22]. It cannot be excluded, however, that these preserved CTL responses are a consequence, rather than a cause, of prolonged asymptomatic survival. Experimental depletion of CD8 T cells in SIV-infected macaques led to the loss of control of acute infection and to increased viral load in chronic infection [23, 24]. Later studies showed, however, that CD4 T cells under these conditions started to proliferate, providing the virus with an excellent opportunity to replicate, which may explain the rise in viral load [25].
Finally, the quality of T cell responses early in infection was not associated with AIDS-free survival [26]. As for neutralizing humoral immunity, this lack of effect of CTLs may be due to viral escape. Escape mutations in even a single epitope may lead to loss of immune control by CTLs [27]. Viruses with CTL escape mutations can be rapidly selected and overtake the viral quasispecies in as little as 6 weeks after first emergence [28]. The rapid selection of these escape variants is in fact the strongest argument that CTLs are effective in vivo. The impact of certain CTL escape mutations on viral fitness [29, 30] was also demonstrated by complete reversal of the escape mutation to the wild-type sequence upon transmission to a new individual [31]. The selective pressure of CTLs is such that at a population level, the virus strains circulating in a population have adapted to the most common human leukocyte antigen (HLA) types in that population [32, 33]. The complex influence of HLA on HIV-1 disease course will be discussed later in the section on host polymorphisms. Mutations outside CTL epitopes have also been described to influence CTL function, by affecting antigen processing and presentation [34].
Cellular immune response: helper T lymphocytes
The functioning of HIV-1-specific CTL depends on the presence of functional CD4 helper T cells. The presence of CD4 helper T cells that proliferate and produce both IL-2 and IFN-γ in response to HIV-specific peptide pools early in infection are not predictive for prolonged AIDS-free survival [35]. However, during the course of infection, the loss of these helper T cells, together with the loss of all functional CD4 T cells, causes a less efficient immune response, thus allowing opportunistic infections to bypass the immune system, finally resulting in disease progression and AIDS. In mouse models it was demonstrated that the presence of CD4 T cells during priming of CTLs is essential for the maintenance of immunological memory after the acute infection phase, whereas their continuous presence was not required for CTL expansion [36, 37]. It can thus be envisioned that with the loss of CD4 T cells during HIV-1 infection no efficient new CD8 T cell memory is generated in later phases of the infection.
Chronic Immune Activation and CD4 T Cell Loss
Depletion of CD4 T cells, which is preceded by a loss of proliferative responses to polyclonal stimuli [38] and recall antigens [39], is one of the major characteristics of HIV-1 infection [40]. The fact that HIV-1 uses CD4 as a cellular entry receptor together with the observation that CD4 T cells are specifically lost in HIV-1 infection led to the logical conclusion that virus-mediated killing of target cells was the main cause for this cell loss. However, the frequency of infected cells in peripheral blood in the chronic phase of infection is too low to account for the ongoing depletion of CD4 T cells by viral infection [41, 42]. Several converging lines of evidence now suggest that hyperactivation of the immune system in response to chronic HIV-1 infection may be the culprit [43]. Hyperactivation is responsible for an increased naive T cell turnover, leading ultimately to the exhaustion of the naive T cell compartment, which then cannot compensate for the enhanced death of memory CD4 T cells into which the activated CD4 T cells mature. Indeed, the level of immune activation was found to be at least as good a predictor of disease progression as the level of viral replication [44, 45]. Additional evidence came from the comparison of SIV infection in sooty mangabeys and African green monkeys on the one hand and in Asian macaque species on the other [46]. SIV infection in Asian macaques generally leads to high virus loads, declining CD4 T cell numbers, and disease within 2 years [47]. Sooty mangabeys and African green monkeys can both be infected with SIV and despite overt viral replication these animals have stable CD4 counts and do not progress to disease [48, 49]. The stable CD4 counts in the presence of high viral load is in line with the assumption that virus-mediated killing has no large effect on total CD4 T cell numbers. Despite the high viral load, these animals show no evidence of hyperactivation of their immune system as observed in SIV-infected macaques and HIV-1-infected humans [49]. This suggests that although the increased turnover of naive T cells in HIV-1-infected individuals is virus driven, the responsiveness is determined by host factors. More recently it was demonstrated that the initial immune response against the virus was similar in both the non-pathogenic and pathogenic macaque model. Interestingly, this initial response was then down-regulated only in the non-pathogenic macaque model but not in the pathogenic SIV macaque model [50–52]. The underlying mechanism for the differential regulation of the immune response needs to be elucidated. It has been suggested that at least two populations of CD4 T cells are important in mediating this immune response: Th17 cells, defined by the secretion of cytokine IL-17, which are thought to be critical in the defense against bacterial and fungal pathogens, and regulatory T cells (Tregs), expressing FoxP3, which are able to induce tolerance against self-antigens and prevent autoimmunity. The pro-inflammatory Th17 cells and immunoregulatory Tregs have antagonistic effector functions; a shift in this balance might be critical in the outcome of HIV disease. Indeed, the loss of Th17/Treg balance was associated with chronic immune activation and pathogenic SIV infection in non-human primates [53]. The mechanisms responsible for selective Th17/Treg imbalance during pathogenic infection are as yet undefined.
CD4 depletion in the gut
In both the SIV macaque model and in HIV-1-infected humans it was demonstrated that already during the phase of primary infection a massive depletion of CD4 T cells occurs in all tissues, including the gut-associated lymphoid tissues (GALT) where more than 80% of T cells reside [54–57]. This depletion of lymphocytes from the GALT occurs within days and seems to be irreversible [58]. It was shown that HIV-1 infection and depletion in the acute phase was not restricted to activated memory cells as also a high frequency of infected resting CD4 T cells could be demonstrated [59, 60]. The unique capacity of HIV-1 to replicate in non-dividing cells is in agreement with this observation [61]. Recently it has been reported that Th17 cells are preferentially lost from the GI tract. Importantly, this loss is observed in HIV-1 infection and pathogenic SIV infection of rhesus macaques, but not in non-pathogenic SIV infection of sooty mangabeys [62, 63].
Viral Factors That Influence Viral Load: Biological Phenotype
HIV-1 can vary with respect to biological properties such as replication rate, cell tropism, co-receptor use, and neutralization sensitivity. The use of different co-receptors by HIV-1 is a general phenomenon for viruses from all different subtypes and circulating recombinant forms (CRFs), although quantitative differences in the prevalence of HIV-1 variants that can use the CXC chemokine receptor 4 (CXCR4-using variants) among subtypes exist. Virus variants with different biological properties dominate in different phases of infection. New infections are generally established by HIV-1 variants that use CD4 and additionally the CC chemokine receptor 5 (CCR5) as a co-receptor (R5 variants) [64–68]. Even if both R5 and CXCR4-using variants are present in the donor, most often only the R5 variants are detected in the recipient [65, 66, 69]. With progression of disease, a shift in the viral quasispecies toward more rapidly replicating T cell-tropic R5 variants is observed [64]. In the natural course of approximately half of HIV-1 subtype B-infected individuals this is associated with the appearance of CXCR4-using HIV-1 variants prior to AIDS diagnosis [70]. CXCR4-using variants are associated with an accelerated CD4 T cell decline and a more rapid disease progression [71, 72]. The accelerated loss of CD4 T cells after appearance of CXCR4-using variants can be explained from the fact that naïve T cells express CXCR4 but not CCR5, while memory cells express both CXCR4 and CCR5 [73]. This co-receptor expression pattern makes naïve T cells a unique target cell population for CXCR4-using variants. Indeed, clonal virus isolation from naïve T cells resulted in predominantly CXCR4-using variants, whereas both R5 and CXCR4-using biological virus clones could be obtained from the memory T cells from the same individuals [74]. It can be envisaged that infection and subsequent virus-mediated killing of naïve T cells by CXCR4-using virus variants directly interferes with T cell ontogeny as the infected and killed naïve T cell will no longer give rise to a daughter cell population. Considering the potential beneficial effect of an expanded target cell population and the limited number of amino acid substitutions in V3 that is sufficient to confer CXCR4-using capability to R5 variants, it is puzzling that CXCR4-using variants emerge prior to AIDS diagnosis in only a proportion of infected individuals [75, 76]. It has been hypothesized that the absence of CXCR4-using variants early in HIV-1 infection may be due to their higher vulnerability to the host adaptive immune responses, in particular neutralizing antibodies. The fact that these variants appear more frequently after the initial decline of CD4 T cells indeed suggests that they may represent a peculiar, congenic form of opportunistic infection. The first appearing CXCR4-using variants are more sensitive to neutralizing antibodies directed against the CD4 binding site than their coexisting R5 variants [77]. Moreover, a conserved neutralization epitope, designated D19, is invariably cryptic in R5 variants of different genetic subtypes, but it is consistently exposed in CXCR4-using variants, rendering such variants sensitive to neutralization by a specific antibody [78].
Evolution of co-receptor use
Phylogenetic analyses of HIV-1 envelope sequences have shown that CXCR4-using variants directly evolve from R5 variants, after which the CXCR4-using and R5 viruses coexist within the same individual [79, 80]. After the first appearance of CXCR4-using variants, both R5 and CXCR4-using variants continuously evolve away from the common ancestor and each other. However, this is only evident for the env gene as CXCR4-using and R5 virus populations cannot be distinguished on the basis of gag sequences due to frequent recombination events outside the env gene [81]. In general, the first CXCR4-using variants can still use CCR5 on primary cells, albeit it far less efficiently [82]. Recent studies have shown that failure of maraviroc, a small molecule CCR5 antagonist, is caused by the presence of R5 variants that can use maraviroc-bound CCR5 for entry [83] or by the presence of CXCR4-using variants, which upon closer inspection were existent pre-therapy [84, 85]. The continued evolution of CXCR4-using and R5 variants is also evident from changes in their biological properties over time. Early CXCR4-using variants are more sensitive to the inhibitory effect of co-receptor antagonists AMD3100 and T22 than late-stage-obtained CXCR4-using variants [82]. In analogy, late-stage R5 variants from individuals who never developed CXCR4-using variants are less sensitive to inhibition by the natural ligand of CCR5, RANTES [86]. The co-receptor inhibitor resistance of R5 and CXCR4-using variants is correlated with the immune status of the host. Although the exact mechanism of resistance remains to be established, the observation is suggestive for selection of HIV-1 variants that use their co-receptor with increasing efficiency as the infection progresses. In a recent study we have observed that viruses with this late-stage phenotype can be transmitted to a new host after which evolution continued [87]. So, while R5 variants are preferentially transmitted over CXCR4-using variants, there does not seem to be a preferential transmission phenotype within R5 variants. The fact that HIV-1 envelope is changing at a population level over calendar time is in line with this observation [88].
Viral accessory genes
Nef
In the SIV macaque model it was demonstrated that inoculation with SIV that lacked the Nef gene resulted in infection but not disease progression [89]. A cohort of individuals in Sydney infected with HIV-1 from the same source and classified as long-term non-progressors, all carried a ΔNef HIV-1 variant [90], although some have ultimately progressed to AIDS-defining events [91]. Multiple Nef activities are known, which are mostly genetically separable and make use of distinct elements located throughout the Nef molecule. Nef alters the intracellular trafficking of important immune molecules, such as class I and II major histocompatibility complex proteins (MHC-I, MHC-II), CD4, and DC-SIGN [92–95]. Down-regulation of MHC-I proteins on the surface of the cell by Nef protects HIV-infected cells from recognition and killing by CTLs, while Nef-dependent CD4 removal enables optimal release of infectious virions. More directly, Nef also triggers apoptotic pathways, affecting survival of bystander CD4 T cells [96]. Furthermore, Nef promotes the induction of cellular transcription factors that can elevate viral replication [97] and Nef intersects with the macrophage CD40L signaling pathway to promote infectivity [98]. Recently it has been reported that Nef can transfer from infected cells into B cells, leading to impaired class switching [99]. Thus, Nef supports viral replication via both direct and indirect mechanisms.
Vif
The HIV-1 protein Vif is essential for viral replication by counteracting the effects of apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like (APOBEC) 3 G and APOBEC3F, mediators of one aspect of the innate immunity, a potent cellular defense system against retroviral infection [100, 101]. APOBEC3F and APOBEC3G are members of the APOBEC superfamily of cytosine deaminases which, in the absence of Vif, are incorporated into the virion. During reverse transcription of the viral genome in a new cell, they deaminate cytidine to uracil, inducing lethal G-to-A hypermutation in the viral DNA [102, 103]. Vif can bind both APOBEC3F and APOBEC3G and redirect it by ubiquitination to degradation in the proteasome, thereby preventing the viral DNA from mutation [104, 105]. More recently it was reported that the expression of truncated or misfolded viral proteins due to APOBEC3G editing enhances the recognition of HIV-1-infected cells by CTLs, linking the innate and adaptive immune responses [106].
Vpr
Monkeys infected with SIV without Vpr function had severely attenuated infections with much lower viral burden and no evidence of disease progression [107], confirming the role of Vpr in viral pathogenesis. Multiple functions of the viral protein Vpr have been reported. First, Vpr has been shown to interact with cellular factors leading to the inhibition of host cell proliferation by a G2 cell cycle arrest of infected cells [108], although the virological role remains unclear. Vpr also has a nuclear localization signal and facilitates nuclear localization of the viral pre-integration complex [109, 110]. By interaction of Vpr with various transcriptional factors on the LTR promoter, the viral protein induces HIV-1 viral gene transcription [111]. Additionally, Vpr causes cell death by inducing apoptosis [112].
Vpu
Two distinct functions have been associated with the viral protein U (Vpu). Vpu down-regulates CD4 cell surface expression by targeting CD4 for degradation in the endoplasmic reticulum of infected cells [113]. Vpu is also known to enhance efficient viral particle release, by antagonizing the action of tetherin [114, 115]. Tetherin is a transmembrane protein that blocks the release of budding HIV-1 virions by directly anchoring the viral particle to the surface of the cell. The retained virions are internalized by endocytosis and subsequently degraded.
Host Factors That Influence HIV-1 Acquisition and Disease Progression
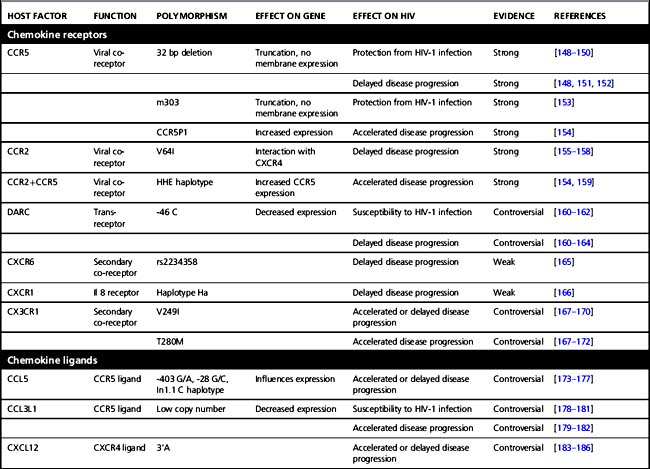
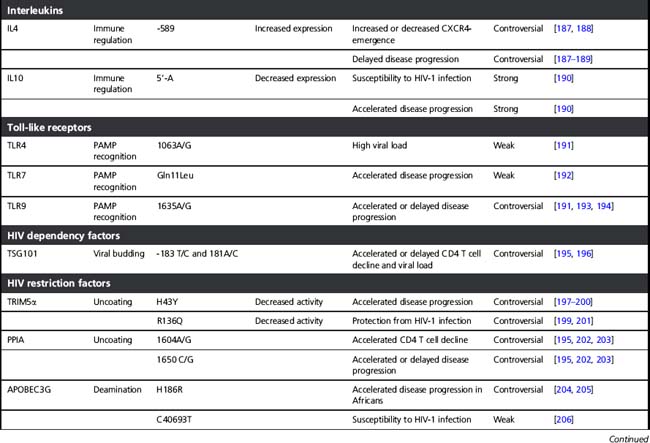
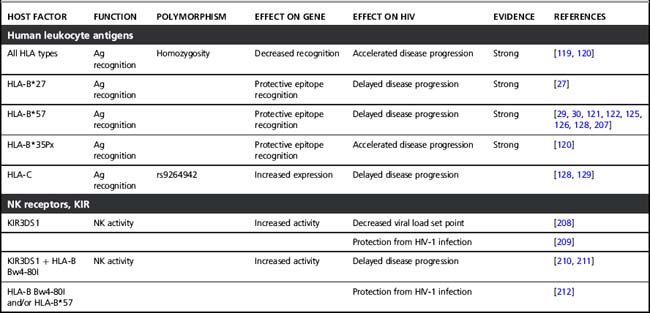
Several host factors that influence the clinical course of HIV-1 infection have been identified. Initially, these host genetic factors were discovered in candidate gene studies (see Table 5.1), in which gene variants that were already known or suspected to play a role in HIV-1 pathogenesis and immune regulation were tested for association with HIV-1 infection and/or disease progression. Examples of these are genes that encode proteins necessary for HIV-1 entry in a cell or for efficient replication and propagation of the virus. In addition, variations in innate and adaptive immune-regulatory genes and in specific viral-restriction genes have been studied for association with HIV-1 disease. Of these, the HLA genes are discussed in more detail below. The variants that were identified in most of these candidate gene studies turned out to have large effects on disease risk, even in small cohorts (see Table 5.1) and most have been reviewed extensively before [116–118]. In the case of CCR5, this has even resulted in the development of new antiviral strategies to block CCR5 in HIV-1-infected individuals, as uninfected individuals without CCR5 function (i.e. those homozygous for the 32 base pair deletion in CCR5) show no overt clinical symptoms.
Table 5.1 Host factors that influence HIV-1 acquisition and disease progression discovered by candidate gene studies
Human leukocyte antigens
The HLA genes map to chromosome 6 and form one of the most polymorphic regions in the human genome. HLA class I loci, HLA-A, B, and C all encode for a large number of different alleles. Each individual expresses two HLA-A, two HLA-B, and two HLA-C alleles on the surface of all their cells. These HLA molecules present viral antigens to CD8 T cells, thereby initiating a cytotoxic T cell response. Due to the large variation that is created by six different HLA molecules, a large diversity of viral peptides that supports potent immunity can be presented. Homozygosity for HLA alleles reduces the repertoire that can be presented to the immune system, thereby limiting the number of epitopes recognized by CTLs. Indeed, homozygosity has been associated with faster disease progression [119, 120]. Moreover, certain class I alleles have been implicated in the variable clinical course of HIV-1 infection. HLA-B*27 [27] and B*57 are consistently associated with effective control of HIV-1 and delayed disease progression, albeit that this association is not absolute [29, 30]. HLA-B*5701 has been associated with long-term non-progression in Caucasian populations [121, 122], while the closely related HLA-B*5703 is associated with delayed disease progression after HIV-1 infection in individuals from African descent [123, 124]. This was confirmed in a GWAS in African Americans [125]. Additionally, HLA-B*57 seems to be associated with control of HIV-1 already early in infection as the prevalence of this allele was significantly lower in individuals with symptomatic acute infection, when compared to a chronically infected population [126]. HLA-B*27 and HLA-B*57 restricted CTL select for HIV-1 variants that have escape mutations in certain epitopes in more conserved regions that come at a relatively high fitness cost to the virus. This is supported by the observation that escape mutations in some epitopes that are restricted by HLA-B*57 immediately revert to wild-type sequences after transmission to a non-HLA-B*57 individual [31]. Interestingly, HIV-1 variants of HLA-B*57-typed individuals with a typical disease course had additional mutations that compensated for the fitness cost associated with the mutations in CTL epitopes. These compensatory mutations were not observed in HLA-B*57-typed long-term non-progressors [29]. Another mechanism for HLA-B*57-related control involves induction of strong CTL responses against the escaped epitopes [127].
HLA-B*35Px, a subgroup of HLA-B*35 based on peptide-binding properties, is associated with a more benign disease course after HIV-1 infection [120], for which the underlying mechanism is not fully clear. While it seems that HLA-B is most frequently implicated in the course of HIV-1 disease, a GWAS revealed genetic variation in the HLA-C gene region to be associated with viral control and slower progression to AIDS [128, 129], confirming earlier data [130]. The protective allele of this polymorphism is associated with high HLA-C cell surface expression, possibly through affecting a 3’UTR miRNA binding site that can degrade or repress translation of the HLA-C gene [129, 131].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


