Chapter 2 The origins and diversification of HIV
The Deep Roots of HIV
AIDS was first recognized in the USA in the early 1980s [1], and the discovery of HIV followed soon after [2]. It is now clear, however, that HIV emerged decades earlier, from naturally infected primates on another continent [3]. Figure 2.1 illustrates the relationships between the different variants of HIV and related viruses that have been discovered in a large number of African monkeys and apes. To date, over 40 species of non-human primates have shown evidence of infection by SIV [4], every one of which is restricted in range to sub-Saharan Africa. Since the primate lentiviruses form a single, distinct clade on the mammalian lentivirus phylogeny, and given that no primates outside of Africa appear to be infected, SIV evidently had its origin in an African monkey at some point sufficiently deep in time to account for its spread throughout most of the continent, and into most of the (catarrhine) primate species there.
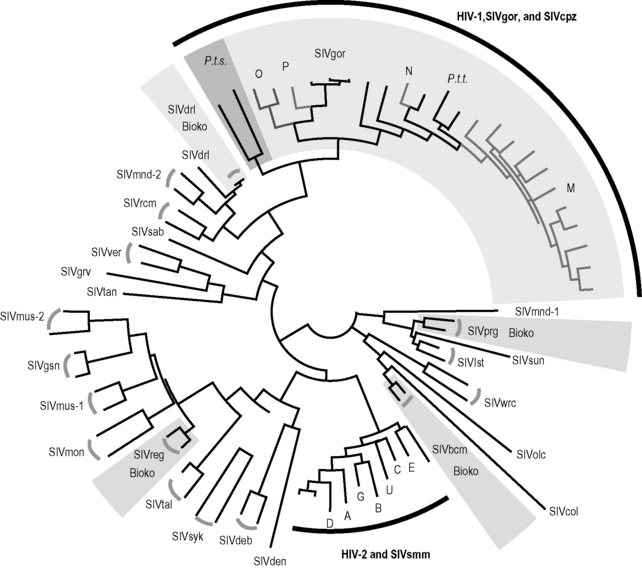
Figure 2.1 Maximum likelihood phylogenetic tree of SIV and HIV partial pol protein sequences.
The sequences were downloaded from the Los Alamos National Laboratories HIV database (http://www.hiv.lanl.gov). The phylogenetic tree was reconstructed under the Jones-Taylor-Thornton (JTT) model of amino acid substitution with gamma distributed with invariant sites. SIVcpz strains recovered from Pan troglodytes troglodytes (P.t.t.) and SIVgor strains from Gorilla gorilla are lightly shaded, while those from P. t. schweinfurthii (P.t.s.) are darkly shaded. The SIV strains isolated from Bioko island are lightly shaded. The abbreviations and species names are as follows: SIVdrl, drill; SIVmnd-1 and SIVmnd-2, mandrill; SIVrcm, red-capped mangabey; SIVsab, green monkey; SIVgrv, grivet; SIVtan, tantalus monkey; SIVver, vervet monkey; SIVmus-1 and SIVmus-2, mustached guenon; SIVgsn, greater spot-nosed monkey; SIVmon, mona monkey; SIVreg, red-eared guenon; SIVtal, talapoin; SIVsyk, Sykes’s monkey; SIVdeb, De Brazza’s monkey; SIVden, Dent’s mona; SIVbcm, black colobus monkey; SIVcol, colobus monkey; SIVolc, olive colobus; SIVwrc, western red colobus; SIVlst, L’Hoest’s monkey; SIVsun, sun-tailed monkey; SIVprg, Preuss’s guenon; SIVsmm, sooty mangabey; SIVcpz, common chimpanzee; SIVgor, gorrilla.
Unlike HIV, natural SIV infections generally cause little illness in their hosts and most are thought to be non-pathogenic. The low pathogenicity has been well documented in African green monkeys and sooty mangabeys. However, a recent breakthrough demonstrated that wild chimpanzees naturally infected with SIV do develop hallmarks of AIDS-like illness; SIV infection, as with HIV-1 and HIV-2 infection, is associated with progressive CD4 cell loss, lymphatic tissue destruction, and premature death [5]. Pan troglodytes schweinfurthii infected with SIV in Gombe National Park in Tanzania have a markedly higher death rate than non-infected animals [5].
The precise nature of the time scale of SIV evolution is still open to question. Initially, comparisons of primate and virus phylogenies led to suggestions that some SIVs have co-diverged with their primate hosts, as the animals split into new species from their common ancestors. However, the pattern of closely related hosts having closely related viruses might be explained not by co-divergence but by cross-species transmission events occurring preferentially between closely related hosts [6]. For example, detailed phylogenetic analysis shows that recent cross-species transmission events, instead of ancient co-divergence, likely underlie the fact that three closely related hominoid primates (human, chimpanzees, and gorillas) harbor closely related lentiviruses [7]. Studies of genes involved in innate immunity, such as APOBEC3G [8], suggest just the sort of mechanism that could generate such a pattern of correspondence even if the viruses and their hosts did not co-diverge.
Hence, even at this advanced stage of the investigation of one of the most medically important pathogens, until recently there has been little agreement on whether its progenitors have been circulating in African primates for millions or just thousands of years. A recent study, however, revealed evidence for several SIV lineages endemic to Bioko Island, Equatorial Guinea. This island was isolated from Africa as sea level rose 10,000 to 12,000 years ago. Notably, each of Bioko’s four SIV lineages is most closely related to a virus circulating in hosts of the same genus on the African mainland rather than to the SIVs of other Bioko species (Fig. 2.1). This phylogeographic approach established that SIV is ancient—at least 32,000 years old [9].
The discovery of an endogenous lentivirus in the genome of the gray mouse lemur (Microcebus murinus) also suggests a time scale of millions of years for primate lentiviruses [10]. On the other hand, molecular clock methods calibrated by using modern sequences make it hard to conceive of dates older than a few thousand years for the SIV MRCA [11, 12]. Extrapolations from the rapid short-term evolutionary rates observed in lentiviruses [13] suggest that, for SIV lineages that diverged more than even a few thousand years ago, the molecular evidence of shared ancestry ought to have become overwritten by a succession of nucleotide substitutions. Clearly, more work is still needed to resolve this conundrum, including developing new models of sequence evolution that incorporate the idiosyncrasies of RNA virus evolution. Nevertheless, it is now very clear that SIV is no newcomer; these viruses have almost certainly been circulating for tens of thousands of years at least, raising the obvious question: what changed within the past hundred years that allowed multiple SIV lineages to successfully establish themselves in the human population?
HIV/SIV Nomenclature
Of the dozens of species with naturally occurring SIV, just three, the common chimpanzee (Pan troglodytes), the gorilla (Gorilla gorilla), and the sooty mangabey (Cercocebus atys), are the putative reservoirs of HIV. HIV type 1 (HIV-1) is the designation given to forms of the human virus linked to SIV from P. troglodytes (SIVcpz) and G. gorilla (SIVgor), while HIV type 2 (HIV-2) denotes human viruses related to the sooty mangabey virus (SIVsmm). Inspection of the SIV/HIV phylogenetic tree shows that, within both SIVcpz/SIVgor and SIVsmm, more than one cross-species transmission event has occurred (Fig. 2.1). The key observation here is that HIV lineages intermingle on the tree with SIV lineages. HIV-1 groups N and M are both more closely related to some of the SIVcpz viruses on the tree than they are to HIV-1 group O, whose lineage branched off the main trunk at an earlier point. In other words, HIV-1 group M shared a most recent common ancestor with a chimpanzee virus, not with HIV-1 group O. HIV-1 groups P and O are most closely related to SIVgor [7, 14]. Likewise, HIV-2 group E is the “sister” group to one SIVsmm strain, and HIV-2 group A is the sister group of another. If there had been only a single transmission from each reservoir species to humans, we would expect the human viruses to fall into single clusters (or monophyletic clades, in the jargon of phylogenetics)—one for HIV-1 and one for HIV-2. This is not the case.
Recombination, whereby genes from separate strains are combined into a new, chimeric viral genome within a dually infected host, complicates these phylogenetic inferences somewhat. For example, HIV-1 group N (or its SIV precursor), though closely related to HIV-1 group M in the pol region (Fig. 2.1), apparently arose from a recombination event [15]: some of its genome is much more distantly related to group M [16]. Such complications aside, the HIV lineages depicted in Fig. 2.1 are thought to have arisen from an independent cross-species transmission, and hence each of these groups is thought to be more closely related to either SIVcpz or SIVsmm than to the other groups in its type.
So, while the different types of HIV denote the different primate reservoirs that have served as sources of human infection, the various groups within each type represent putative independent introductions from primate to human. Within HIV-1, at least four such events are inferred, giving rise to HIV-1 groups M, O [17], N [15], and P [14]. Within HIV-2, eight independent transmissions are indicated, corresponding to HIV-2 groups A through H [3, 18]. Earlier studies of HIV-2 used the term subtype for these lineages, but this usage has given way to the use of the term group in order to bring HIV-2 nomenclature in line with the more widely cited (if slightly less logical) HIV-1 conventions [18]. The goal of the change was to emphasize the biological parallel between the different lineages of HIV-1 and HIV-2 that arose via unique zoonotic origins. In this sense, HIV nomenclature reflects rather well the evolutionary processes driving observed patterns of genetic diversity.
The term subtype, in turn, is used within HIV-1 group M. Each of the branches in the M group in Fig. 2.1 represents one of the recognized subtypes (A to D, F to H, J, K). Different subtypes dominate in different regions. For example, subtype B accounts for most infections in Europe and the Americas, subtype C predominates in southern African countries like South Africa, as well as in India, and subtype D is common in east Africa. The unfortunate fact that subtypes are nested within groups, rather than types, in this naming scheme, owes to the fact that the use of the term predates the discovery of HIV-1 group O, at which point the new designation of group had to be wedged between type and subtype. The system of naming HIVs and SIVs has thus evolved over time as the full diversity of natural SIV infections in African primates was revealed, and as new, distinct lineages of HIV have been discovered.
As hinted above, recombination plays a large role in the evolution of both SIV and HIV [19]. The existence of clear recombinants, with genomes that are mosaics of distinct lineages, has led to the introduction of a formal system for recognizing them [20]. These circulating recombinant forms (CRFs) represent virus populations that have diversified from a single, ancestral strain generated by recombination between two or more of the recognized M group subtypes [20]. The “missing” subtypes, E and I, have been re-classified as CRFs after detailed analysis of their genomes revealed that they had recombinant origins [20]. Some CRFs are the dominant HIV-1 group M strain in some locales (e.g. CRF01 in Thailand, CRF02 in Nigeria).
There is also abundant evidence of recombination among the SIVs of different primate species, and even between HIV-1 groups M and O [21]. Most notably, the progenitor of HIV-1, SIVcpz, turns out to be a recombinant between the SIVs of red-capped mangabeys and greater spot-nosed monkeys, two prey species of the chimpanzee [22]. All strains of HIV-1 are thus ultimately recombinant in origin, their genomes a mosaic of two monkey viruses, trafficked through an ape intermediary.
Where did HIV Enter the Human Population?
Using the geographic distributions of the primate species that have spawned HIV variants it has been possible to infer, with remarkable precision, the specific areas in Africa where the various HIV-1 and HIV-2 groups originated. HIV-2 was the first to give up its secrets, and by the early 1990s it was clear that different groups of HIV-2 were independently derived from SIVsmm, the SIV variant endemic in the sooty mangabey monkeys of West Africa [23]. HIV-2 is endemic to the same region, and is only rarely detected elsewhere. Although HIV-2 accounts for relatively few infections compared with HIV-1, many more cross-species transmissions involving this virus have been detected, with eight groups (A–H) currently recognized. Only two of these, groups A and B, appear to have established themselves as endemic human infections. In all the other cases, the “group” is in fact composed of a single patient infected with an HIV-2 variant that is sufficiently genetically divergent from the others that it was likely acquired independently. Some or all of these may represent evolutionary “dead ends [24].” In some cases, the human virus bears a surprisingly close resemblance to SIVsmm, infecting free-living or pet sooty mangabeys from the same local area [24, 25], strong evidence of repeated independent cross-species transmission.
The geographical origins of HIV-1 have taken longer to piece together, but it now seems clear that all four HIV-1 groups, as well as SIVgor, form a monophyletic cluster with SIVcpz from P. troglodytes troglodytes, the “central” chimpanzee, whose range encompasses southern Cameroon, Central African Republic, Equatorial Guinea, Gabon, and the Republic of Congo (Congo-Brazzaville) [3, 16, 26, 27]. Although both the central chimpanzee and the “eastern” subspecies, P. t. schweinfurthii, are naturally infected with SIVcpz, the viruses recovered from the two chimpanzee lineages form distinct clades on the SIV phylogenetic tree (Fig. 2.1), indicating that they have been evolving in isolation for a considerable time.
While two instances of cross-species transmission from chimpanzees to humans (HIV-1 groups M and N) are unequivocally phylogenetically linked to the SIVcpz of the P. t. troglodytes chimpanzee, HIV-1 groups O and P fall within the radiation of SIVcpz strains but are mostly closed to SIVgor of G. gorilla (Fig. 2.1). It remains unclear whether gorillas were the immediate source of either or both of HIV-1 groups O and P. No known variant of HIV-1 has emerged from the eastern chimpanzee (dark shading in Fig. 2.1). HIV-1 groups N, O, and P are all endemic to Cameroon, within the range of P. t. troglodytes, and none have spread substantially beyond this presumptive region of origin. Group O exhibits about 0.4% prevalence in Cameroon [28], group N is exceedingly rare, with less than 10 infected individuals identified to date [29], and group P infections are also extremely rare, accounting for only 0.06% of HIV infections [30]. Despite the relative rarity of groups N and O, their pathogenic profile appears indistinguishable from group M’s [28], an indication that pathogenic potential and epidemic behavior of AIDS viruses are not necessarily coupled [18]. Since group P was identified only recently, little is known on its pathogenic profile.
Given the very different properties of the four groups, M, O, N, and P, in terms of rate of spread through human host populations, it is tempting to speculate that their pathogenic properties may owe more to their common genetic heritage, as close relatives descended from SIVcpz, than to convergent evolutionary trajectories once they entered humans. The discovery of the first rare variant of HIV-2 known to cause immunosuppression lends support to this notion [18], but future studies will be required to further clarify the ground rules of the evolution of HIV pathogenicity.
The geographical source of the main group of HIV-1 has been somewhat obscured by its global spread, but the available evidence links it to the same region. Group M falls soundly among the diverse viruses of the P. t. troglodytes chimpanzees—powerful evidence that it emerged from within their range [16]. By screening non-invasively collected fecal samples from the region, Beatrice Hahn and colleagues have identified several closely related SIVcpz strains from wild-living Cameroonian chimpanzees, viruses that are remarkably similar to group M, and which form a well-supported cluster with it on phylogenetic trees. These analyses pinpoint the probable source of the viruses that gave rise to the HIV-1 group M pandemic as being chimpanzees in southeastern Cameroon [31]. From there, the virus likely made its way, perhaps diffusing along the Sangha River and then down the Congo River, to Kinshasa [32]. While the fuse was evidently lit in rural southeastern Cameroon, the truly explosive growth of the pandemic was likely linked to its arrival in the region’s largest city.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


