(tah-KROH-lih-mus)
Prograf
Immunosuppressant
Usual dose
See Precautions. IV route used for patients unable to take oral medications; risk of anaphylaxis increased with IV administration. Transfer to oral therapy as soon as tolerated, usually within 2 to 3 days.
For all indications:
Dose regimens vary among transplant centers and approved or investigational use (range 0.01 to 0.05 mg/kg/day). Begin no sooner than 6 hours after transplantation in liver and heart transplant patients. In kidney transplant patients, initial dose may be administered within 24 hours of transplantation but should be delayed until renal function has recovered. Adults usually receive doses at the lower end of the range. Pediatric patients may require doses at the upper end of the range. Individualized adjustment based on clinical assessment of rejection or patients’ tolerance is imperative and may be required on a daily basis. Adjunctive adrenal corticosteroid therapy early posttransplant is recommended. Initiate oral tacrolimus therapy as soon as feasible. Oral doses vary with specific organ transplant and in adults and pediatric patients (see literature). Total dose in mg/kg/day is given in equally divided doses every 12 hours. Begin 8 to 12 hours after IV tacrolimus is discontinued. Lower doses may be sufficient for maintenance therapy.
Prophylaxis of organ rejection in heart transplant:
The recommended starting dose is 0.01 mg/kg/day as a continuous infusion. Used in combination with azathioprine or mycophenolate (CellCept) in addition to adrenal corticosteroids. See all comments and protocol under For All Indications above.
Prophylaxis of organ rejection in kidney transplant:
The recommended starting dose is 0.03 to 0.05 mg/kg/day as a continuous infusion. Used in combination with azathioprine or mycophenolate (CellCept) in addition to adrenal corticosteroids. See all comments and protocol under For All Indications above.
Prophylaxis of organ rejection in liver transplant:
The recommended starting dose is 0.03 to 0.05 mg/kg/day as a continuous infusion. Used in combination with adrenal corticosteroids. See all comments and protocol under For All Indications above.
Pediatric dose
Has been used successfully in pediatric patients up to 16 years of age in liver transplants. See Usual Dose for mg/kg/day dose recommendations. Studies indicate that higher doses may be required to maintain blood trough concentrations similar to adults. Experience in pediatric heart and kidney transplant patients is limited. See Dilution and Maternal/Child.
Dose adjustments
Dosing should be titrated based on clinical assessments of rejection and tolerability. ■ Use lowest dosing range initially for patients with impaired renal or hepatic function (pretransplant or posttransplant). Nephrotoxicity may be increased and further reductions may be required. Half-life prolonged and clearance decreased in patients with severe hepatic impairment (Child-Pugh greater than or equal to 10). Dose reduction may be required. ■ In kidney transplant patients with postoperative oliguria, the initial dose of tacrolimus should be administered no sooner than 6 hours and within 24 hours of transplantation, but it may be delayed until renal function shows evidence of recovery. ■ Lower doses may be appropriate for maintenance. ■ Black patients who have undergone a kidney transplant may require a higher dose to obtain trough concentrations comparable to those seen in white patients. ■ See Monitor and Drug/Lab Interactions.
Dilution
Specific techniques required; see precautions.
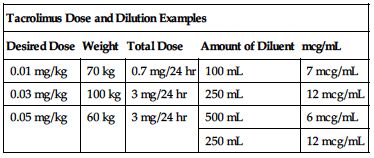
Available as a 5-mg/mL solution in a 1-mL ampule. A 24-hour dose must be diluted with an appropriate amount of NS or D5W. Desired concentration is between 4 and 20 mcg/mL. May leach phthalate from polyvinylchloride containers; mix in glass or polyethylene infusion bottles. Use PVC-free IV tubing in pediatric patients. The following chart provides some dose and dilution examples.
| Tacrolimus Dose and Dilution Examples | ||||
| Desired Dose | Weight | Total Dose | Amount of Diluent | mcg/mL |
| 0.01 mg/kg | 70 kg | 0.7 mg/24 hr | 100 mL | 7 mcg/mL |
| 0.03 mg/kg | 100 kg | 3 mg/24 hr | 250 mL | 12 mcg/mL |
| 0.05 mg/kg | 60 kg | 3 mg/24 hr | 500 mL | 6 mcg/mL |
| 250 mL | 12 mcg/mL | |||
Storage:
Store between 5° and 25° C (41° and 77° F) before dilution. Discard diluted solution in 24 hours.
Compatibility (underline indicates conflicting compatibility information)
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
Manufacturer states, “Should not be mixed with solutions of pH 9 or greater (e.g., acyclovir [Zovirax], ganciclovir [Cytovene IV]).” See Dilution.
One source suggests the following compatibilities:
Y-site:
Aminophylline, amphotericin B (conventional), ampicillin, ampicillin/sulbactam (Unasyn), anidulafungin (Eraxis), benztropine (Cogentin), calcium gluconate, caspofungin (Cancidas), cefazolin (Ancef), cefotetan, ceftazidime (Fortaz), ceftriaxone (Rocephin), cefuroxime (Zinacef), chloramphenicol (Chloromycetin), ciprofloxacin (Cipro IV), clindamycin (Cleocin), dexamethasone (Decadron), digoxin (Lanoxin), diphenhydramine (Benadryl), dobutamine, dopamine, doripenem (Doribax), doxycycline, erythromycin (Erythrocin), esmolol (Brevibloc), fluconazole (Diflucan), furosemide (Lasix), gentamicin, heparin, hydrocortisone sodium succinate (Solu-Cortef), hydromorphone (Dilaudid), imipenem-cilastatin (Primaxin), insulin (regular), isoproterenol (Isuprel), leucovorin calcium, lorazepam (Ativan), methylprednisolone (Solu-Medrol), metoclopramide (Reglan), metronidazole (Flagyl IV), micafungin (Mycamine), morphine, multivitamins (M.V.I.), mycophenolate (CellCept IV), nitroglycerin IV, nitroprusside sodium, oxacillin (Bactocill), penicillin G potassium, phenytoin (Dilantin), potassium chloride (KCl), propranolol, ranitidine (Zantac), sodium bicarbonate, sulfamethoxazole/trimethoprim, tobramycin, vancomycin.
Rate of administration
A single dose properly diluted and equally distributed over 24 hours as a continuous infusion. Use of a metriset (60 gtt/min) or infusion pump suggested.
Actions
A potent immunosuppressive agent. Prolongs survival of allogeneic heart, kidney, and liver transplants. Inhibition of T-lymphocyte activation results in immunosuppression. Highly protein bound. Metabolized primarily by the P450 enzyme system (CYP3A). Half-life ranges from 32 to 55 hours. Primarily excreted in feces. Minimal excretion in urine. Crosses the placental barrier. Secreted in breast milk.
Indications and uses
Prophylaxis of organ rejection in allogeneic heart, kidney, and liver transplants in conjunction with adrenal corticosteroids. In heart and kidney transplant patients, concomitant use with azathioprine or mycophenolate is recommended in addition to the adrenal corticosteroids.
Limitations of use:
Should not be used simultaneously with cyclosporine. ■ Should be reserved for patients unable to take oral tacrolimus. ■ Use with sirolimus (Rapamune) is not recommended in liver and heart transplant patients. Safety and effectiveness of tacrolimus with sirolimus in kidney transplant patients have not been established.
Unlabeled uses:
Treatment of autoimmune diseases (i.e., rheumatoid arthritis). ■ Prevention and treatment of acute graft-versus-host disease.
Contraindications
Hypersensitivity to tacrolimus or polyoxyl 60 hydrogenated castor oil.
Precautions
Follow guidelines for handling hazardous agents. See Appendix A, p. 1331. ■ For IV use only. ■ Oral dosing preferred; begin as soon as feasible. Risk of anaphylaxis is increased by IV route versus oral route; use caution. Reserve for patients unable to take oral medication. ■ Usually administered in the hospital by or under the direction of a physician experienced in immunosuppressive therapy and management of organ transplant patients. ■ Adequate laboratory and supportive medical resources must be available. ■ Use caution in impaired renal function; increases in SCr may require dose reduction or use of an alternate immunosuppressant. ■ Use caution in severe hepatic dysfunction (mean Pugh score: greater than 10); clearance decreased. ■ Can cause nephrotoxicity and neurotoxicity, particularly at higher doses; see Monitor. ■ Posterior reversible encephalopathy syndrome (PRES), delirium, and coma have been reported; see Monitor. ■ Hypertension is a common adverse effect and may require hypertensive therapy; see Drug/Lab Interactions. ■ Use combination immunosuppressant therapy with caution; may oversuppress the immune system and increase susceptibility to infection. ■ Immunosuppressed patients are at increased risk for bacterial, viral, fungal, and protozoal infections, including opportunistic infections. ■ Opportunistic infections may include latent viral infections such as polyoma virus infections. Polyoma virus infections may result in polyoma virus–associated nephropathy (PVAN) and JC virus–associated progressive multifocal leukoencephalopathy (PML). PVAN is associated with serious outcomes, including deteriorating renal function and kidney graft loss. PML is a serious progressive neurologic disorder caused by infection of the CNS by JC virus, a member of the papovavirus family. It typically occurs in immunocompromised patients. PML is rare but may result in irreversible neurologic deterioration and death, and there is no known effective treatment. Hemiparesis, apathy, confusion, cognitive deficiencies, and ataxia are the most commonly observed clinical signs. ■ Patients receiving immunosuppression are at increased risk for developing cytomegalovirus (CMV) viremia and CMV disease. ■ Antiviral prophylaxis (e.g., ganciclovir, valganciclovir) may be advisable in some patients. ■ Concurrent use with sirolimus (Rapamune) is not recommended in liver or heart transplant patients. Regimen is associated with an increased risk of wound healing complications, renal function impairment, and insulin-dependent, posttransplant diabetes mellitus in heart transplant patients; regimen is associated with excess mortality, graft loss, and hepatic artery thrombosis in liver transplant patients. The safety and efficacy of tacrolimus and sirolimus have not been established in kidney transplant patients. ■ Post–liver transplant patients experiencing hepatic impairment may be at increased risk of developing renal insufficiency related to elevated tacrolimus concentrations. ■ May cause lymphomas and other malignancies (especially of skin). Risk appears to be related to intensity and duration of immunosuppression. Has also been associated with a posttransplant lymphoproliferative disorder (PTLD), usually related to Epstein-Barr virus infection. ■ New-onset diabetes mellitus has been reported in tacrolimus-treated heart, kidney, and liver transplant patients. May be reversible over 1 to 2 years in some patients. Black and Hispanic kidney transplant patients may be at an increased risk; see Monitor. ■ Myocardial hypertrophy has been reported in pediatric patients and adults. Reversible in most cases with dose reduction or discontinuation of tacrolimus. ■ May prolong the QT/QTc interval and cause torsades de pointes. Avoid use in patients with congenital long QT syndrome. Patients with CHF, bradyarrhythmias, or electrolyte abnormalities (e.g., hypokalemia, hypocalcemia, hypomagnesemia) and patients taking medicinal products that may lead to QT prolongation may be at increased risk; see Monitor and Drug/Lab Interactions. ■ Pure red cell aplasia (PRCA) has been reported. ■ GI perforation has been reported. All reported cases were considered to be a complication of transplant surgery or accompanied by infection, diverticulum, or malignant neoplasm. ■ See Drug/Lab Interactions.
Monitor:
Obtain baseline CBC, differential, platelets, electrolytes, fasting glucose, BUN, SCr, and liver function tests. Monitor regularly during therapy. ■ Contains a castor oil derivative and alcohol; observe continuously for signs of a hypersensitivity reaction (e.g., acute respiratory distress syndrome [ARDS], dyspnea, pruritus, rash) for the first 30 minutes of the infusion and frequently thereafter. A source of oxygen and epinephrine must always be available. ■ Monitor urine output and SCr carefully. Overt nephrotoxicity occurs more frequently early after transplant. Nephrotoxicity may be acute or chronic. Acute nephrotoxicity is characterized by increasing SCr, hyperkalemia, and/or a decrease in urine output and is usually reversible. Chronic nephrotoxicity is usually progressive and is associated with increased SCr, decreased kidney graft life, and characteristic histologic changes on renal biopsy. Consider changing to another immunosuppressive therapy in patients with persistent elevations of SCr who are unresponsive to dose adjustments. ■ Monitor for S/S of neurotoxicity (e.g., changes in motor or sensory function, changes in mental status, coma, delirium, headache, paresthesias, tremors, seizures). ■ Monitor for S/S of PRES (e.g., altered mental status, headache, hypertension, seizures, and visual disturbances). Diagnosis may be confirmed by MRI. Maintain BP control, and an immediate decrease of immunosuppression is advised. Symptoms have reversed when immunosuppression is reduced or discontinued. ■ Monitor for S/S of PML; apathy, ataxia, cognitive deficiencies, confusion, and hemiparesis are the most commonly observed clinical signs. Consultation with a neurologist may be indicated. ■ Tacrolimus whole blood trough concentrations in conjunction with other laboratory parameters and clinical parameters are used to evaluate rejection, toxicity, need for dose reduction, and patient compliance. Risk of toxicity (e.g., nephrotoxicity, neurotoxicity, posttransplant diabetes mellitus) is increased with higher trough concentrations. Whole blood median trough concentrations may vary considerably during the first week but then stabilize. Most patients are stable when trough whole blood concentrations are between 5 and 20 ng/mL depending on indication and length of time since the transplant. The trough concentrations described above and in the manufacturer’s prescribing information pertain only to oral administration of tacrolimus. Monitoring tacrolimus concentrations during a continuous IV infusion of tacrolimus may have some utility; however, the observed concentrations will not represent exposures comparable to those estimated by the trough concentrations observed in patients on oral therapy. ■ Monitor all parameters to evaluate possibility of organ rejection. ■ Monitor BP; antihypertensives may be indicated; see Drug/Lab Interactions. ■ Monitor electrolyte concentrations. May cause hyperkalemia or hypomagnesemia. ■ In patients with CHF, bradyarrhythmias, or electrolyte abnormalities (e.g., hypokalemia, hypocalcemia, hypomagnesemia) and in patients taking certain antiarrhythmics or other medicinal products that may lead to QT prolongation, consider obtaining an electrocardiogram and monitoring electrolytes periodically during treatment. ■ May cause hyperglycemia. Monitor carefully; treatment may be required. ■ Observe for signs of infection (e.g., fever, sore throat, tiredness) or unusual bleeding or bruising. ■ Consider echocardiographic evaluation in patients who develop renal failure or clinical manifestations of ventricular dysfunction. Consider dose reduction or discontinuation of tacrolimus if myocardial hypertrophy is diagnosed. ■ See Precautions and Drug/Lab Interactions.
Patient education:
Review manufacturer’s medication guide. ■ Nonhormonal birth control preferred to oral contraceptives to reduce complications of drug interactions. ■ Emphasize need for frequent routine lab work; compliance imperative. ■ Interacts with many medications. Discuss any changes in drug regimen (prescription or nonprescription) with doctor or pharmacist. ■ Promptly report any side effects (e.g., hypertension, nephrotoxicity, neurotoxicity, S/S of infection). ■ Review S/S of diabetes mellitus. ■ Inform patient of increased risk of neoplasia, including malignant skin changes. Limit exposure to sunlight and ultraviolet light. ■ See Appendix D, p. 1333.
Maternal/child:
Category C: use only if necessary; benefits must outweigh risk to fetus. Has been associated with hyperkalemia and renal dysfunction in the fetus. ■ Discontinue breast-feeding. ■ Safety and effectiveness for use in pediatric kidney and heart transplant patients not established. ■ Appears to be an increased risk for lymphoproliferative disorder (LPD) and primary Epstein-Barr virus infection in immunosuppressed pediatric patients.
Elderly:
Consider age-related organ impairment.
Drug/lab interactions
Used concurrently with adrenocorticosteroids and azathioprine or mycophenolate (CellCept). Do not use simultaneously with cyclosporine (Sandimmune). If a change of immunosuppressants is indicated (tacrolimus to cyclosporine or cyclosporine to tacrolimus), avoid additive nephrotoxicity by waiting for at least 24 hours before starting the alternate drug. If elevated blood concentrations are present, further extend the interval between the two drugs. ■ Concurrent use with sirolimus (Rapamune) is not recommended; see Precautions. ■ Coadministration with strong CYP3A4 inhibitors (e.g., azole antifungal agents [e.g., itraconazole (Sporanox), ketoconazole (Nizoral), voriconazole (VFEND)], boceprevir [Victrelis], clarithromycin [Biaxin], ritonavir [Norvir], telaprevir [Incivek]) and strong inducers (e.g., rifampin [Rifadin], rifabutin [Mycobutin]), including those that prolong the QT interval (e.g., azole antifungals, clarithromycin), is not recommended without adjustments in tacrolimus dosing and close monitoring of tacrolimus whole blood trough concentration and associated side effects. ■ Concurrent use with other drugs that prolong the QT interval may require dose adjustment and monitoring of the QT interval and trough concentrations. Amiodarone (Nexterone) has been reported to increase tacrolimus whole blood concentration with or without concurrent QT prolongation. ■ Use extreme caution with other nephrotoxic agents (e.g., aminoglycosides [gentamicin, tobramycin], amphotericin B [conventional], cisplatin, ganciclovir [Cytovene], nucleotide reverse transcriptase inhibitors [e.g., tenofovir (Viread)], and protease inhibitors [e.g., ritonavir (Norvir), indinavir (Crixivan)]). ■ Do not use potassium-sparing diuretics (e.g., spironolactone [Aldactone]); increases risk of hyperkalemia. Other agents associated with hyperkalemia (e.g., ACE inhibitors [enalapril (Vasotec)] and angiotensin receptor blockers [e.g., losartan (Cozaar)]) should be used with caution. ■ Calcium channel blockers (e.g., diltiazem [Cardizem], nicardipine [Cardene], nifedipine [Procardia], verapamil), bromocriptine (Parlodel), chloramphenicol (Chloromycetin), cimetidine (Tagamet), danazol (Cyclomen), erythromycin, estradiol (Estrace), herbal products containing schisandra sphenanthera extracts, lansoprazole (Prevacid), methylprednisone (Solu-Medrol), metoclopramide (Reglan), metronidazole (Flagyl), omeprazole (Prilosec), protease inhibitors (e.g., ritonavir [Norvir], saquinavir [Invirase]), theophylline, and troleandomycin (TAO) may inhibit the P450 enzyme system and increase tacrolimus blood levels, increasing toxicity potential. Tacrolimus toxicity may also be increased if given concurrently with nefazodone. ■ Avoid concomitant use with nelfinavir unless benefits outweigh risks. ■ When initiating therapy with posaconazole or voriconazole in patients already receiving tacrolimus, decrease tacrolimus dose to one third of the original dose. Adjust subsequent tacrolimus doses based on tacrolimus whole blood concentrations. ■ Anticonvulsants (e.g., carbamazepine [Tegretol], phenobarbital, phenytoin [Dilantin]), caspofungin (Cancidas), rifamycins (e.g., rifabutin [Mycobutin], rifampin [Rifadin]), and St. John’s wort may induce the P450 enzyme system, decreasing effectiveness and leading to decreased tacrolimus blood levels and organ rejection. ■ Tacrolimus may increase serum concentrations of phenytoin (Dilantin). ■ Grapefruit juice may affect certain enzymes of the P450 system and should be avoided. ■ Do not use live virus vaccines in patients receiving tacrolimus.
Side effects
Heart transplant:
Abnormal renal function, anemia, bronchitis, CMV infection, diabetes mellitus, hyperglycemia, hyperlipidemia, hypertension, infection, leukopenia, pericardial effusion, tremor, and urinary tract infection are most common.
Kidney transplant:
Abdominal pain, abnormal renal function, constipation, diarrhea, headache, hypertension, infection, insomnia, nausea, and tremor are most common.
Liver transplant:
Abdominal pain, abnormal renal function, anemia, asthenia, diarrhea, fever, headache, hyperglycemia, hyperkalemia, hypertension, hypomagnesemia, insomnia, nausea, pain, paresthesia, and tremor are most common. The above side effects are most common by diagnosis, but any of them may occur with any patient receiving a heart, liver, or kidney transplant. Side effects occur in a majority of patients and are more pronounced at higher doses. May improve somewhat over time. Nephrotoxicity (abnormal renal function with increased SCr and BUN, oliguria) and neurotoxicity (delirium, headache, insomnia, paresthesia, tremor, seizures) may be dose limiting. Abnormal ECG, abnormal liver function tests, acute kidney failure, albuminuria, anorexia, arrhythmias, ascites, atelectasis, back pain, blood dyscrasias, CHF, coma, cushingoid features, dyspnea, gastroenteritis, glycosuria, hearing loss (including deafness), hemolytic-uremic syndrome, hypokalemia, hypophosphatemia, impaired wound healing, leukocytosis, leukoencephalopathy, myocardial hypertrophy, neuralgia, pancreatitis, peripheral edema, peritonitis, photosensitivity, pleural effusion, pruritus, pulmonary edema, QT prolongation, rash, seizures, Stevens-Johnson syndrome, thrombocytopenia, thrombophlebitis, torsades de pointes, vertigo, and vomiting have also been reported. Many other side effects have occurred in fewer than 3% of patients.
Post-marketing:
Acute respiratory distress syndrome, agranulocytosis, cardiac arrhythmia, cerebral infarction, DIC, enterocolitis, gastroesophageal reflux disease, GI perforation, hemolytic anemia, hepatotoxicity, interstitial lung disease, posterior reversible encephalopathy syndrome (PRES), PRCA, primary graft dysfunction, progressive multifocal leukoencephalopathy (PML), and PVAN nephropathy are some of the many additional side effects reported.
Antidote
Notify the physician of all side effects. Most will be treated symptomatically. Tacrolimus may be decreased or discontinued or alternate immunosuppressive agents substituted. Nephrotoxicity, neurotoxicity, or hematopoietic depression may require temporary reduction of dose or discontinuation of therapy. Reduction in immunosuppression should be considered for patients who develop evidence of PVAN, PML, or CMV viremia and/or CMV disease. However, the risk that reduced immunosuppression represents to the functioning allograft must also be considered. If S/S of PRES occur, maintain BP control, and an immediate decrease of immunosuppression is advised. Consider discontinuing tacrolimus if PRCA is diagnosed. Dialysis is not effective in overdose. Discontinue immediately if anaphylaxis occurs and treat with oxygen, epinephrine, corticosteroids, and/or antihistamines (e.g., diphenhydramine [Benadryl]). Resuscitate as necessary.
Taliglucerase alfa
(tal-e-GLUE-sir-ace AL-fa)
Elelyso
Enzyme replenisher
(glucocerebrosidase)
pH 6
Usual dose
Treatment-naïve adults and pediatric patients 4 years of age and older:
60 units/kg of body weight administered once every 2 weeks as a 60- to 120-minute IV infusion.
Patients switching from imiglucerase:
Patients currently being treated with imiglucerase for Type 1 Gaucher disease can be switched to taliglucerase alfa. Patients on a stable dose of imiglucerase may transfer to the same unit/kg dose of taliglucerase alfa. Administer dose every other week as a 60- to 120-minute IV infusion.
Dose adjustments
Patients switching from imiglucerase:
Dose may increase or decrease based on achievement of each patient’s therapeutic goals. Optimal goal is to establish the lowest dose that is effective in maintaining control of the disease and preventing a recurrence of symptoms for each patient.
Dilution
Available as a lyophilized powder in 200-unit vials. Determine the number of vials required using the following calculations:
Weight in kg × Dose/kg desired ÷ 200 (units/vial) = # of vials required
A 60-kg man requiring 60 units/kg would require 3,600 units. The number of vials required would be 18. If necessary, round up to the next whole vial. After patient is weighed and appropriate dose is calculated, remove sufficient vials from refrigerator. Do not leave vials at RT for longer than 24 hours before reconstitution.
Each 200-unit vial must be reconstituted with 5.1 mL of SWFI to yield a reconstituted product with a concentration of 40 units/mL and an extractable volume of 5 mL. Gently swirl to mix the solution. Do not shake. Withdraw exactly 5 mL (40 units/mL) from each 200-unit vial. A total dose must be further diluted with NS to a total volume of 100 to 200 mL. For pediatric patients, a final volume of 100 to 120 mL should be used. For adult patients, a final volume of 130 to 150 mL may be used. However, if the volume of the reconstituted product alone is equal to or greater than 130 to 150 mL, then the final volume should not exceed 200 mL. Mix gently; do not shake. Do not use if discolored or opaque or if particulate matter is present. After dilution, slight flocculation (thin translucent fibers) may occur.
Filters:
Should be administered through an in-line, low–protein-binding, 0.2-micron filter.
Storage:
Before use, refrigerate at 2° to 8° C (36° to 46° F). Protect from light. Do not freeze. Do not use after expiration date on bottle. Contains no preservative. Following reconstitution, immediate use is preferred; however, reconstituted and/or diluted vials may be refrigerated for up to 24 hours at 2° to 8° C (36° to 46° F) protected from light. Reconstituted vials are stable for up to 4 hours at 20° to 25° C (68° to 77° F) without protection from light. Store reconstituted and/or diluted product for no more than 24 hours protected from light. Do not freeze, and discard any unused product.
Compatibility
Specific information not available. Consider specific use; consult pharmacist.
Rate of administration
Use of an infusion set with an in-line, low–protein-binding, 0.2-micron filter is required. Use of an infusion pump is helpful. Flush the IV line with NS at the end of the infusion to ensure the total dose is received. Administer as an infusion over 60 to 120 minutes as outlined in the following paragraphs.
Pediatric patients:
Begin with a rate of 1 mL/min. If the patient tolerates the initial infusion rate, it may be increased to 2 mL/min. Total time of the infusion should be no less than 1 hour.
Adult patients:
Begin with a rate of 1.2 mL/min. If the patient tolerates the initial infusion rate, it may be increased to 2.2 mL/min. Total time of the infusion should be no less than 1 hour. Reduction in the rate of infusion may be helpful in the event of side effects or infusion reactions.
Actions
A hydrolytic lysosomal glucocerebroside-specific enzyme. It is produced by recombinant DNA technology using plant cell culture (carrot). An active form of the lysosomal enzyme β-glucocerebrosidase. It catalyzes the hydrolysis of glucocerebroside to glucose and ceramide, reducing the amount of accumulated glucocerebroside normally seen in patients with Gaucher disease. In clinical trials, spleen and liver size were reduced, and anemia and thrombocytopenia were improved. Median terminal half-life was 32.5 minutes in pediatric patients and 28.7 minutes in adult patients. No significant accumulation was noted with repeated dosing.
Indications and uses
Treatment of patients with a confirmed diagnosis of Type 1 Gaucher disease.
Contraindications
Manufacturer states, “None.”
Precautions
Should be used under the direction of a physician knowledgeable in the management of Gaucher disease and administered in a facility with adequate diagnostic and treatment facilities to monitor the patient and respond to any medical emergency. ■ Hypersensitivity reactions, including anaphylaxis, have occurred; see Monitor and Antidote. ■ A therapeutic protein product, patients have developed IgG antidrug antibodies (ADA) to taliglucerase alfa. The relationship between ADA and hypersensitivity reactions is not fully understood. Monitoring for ADA to taliglucerase alfa may be useful in ADA-positive patients or in patients who have experienced hypersensitivity reactions to taliglucerase alfa. Neutralizing antibodies have also been detected in some patients receiving taliglucerase alfa. Clinical relevance as it relates to therapeutic effect is unclear.
Monitor:
Obtain weight before each dose (used to calculate dose). Monitor vital signs. ■ Observe patient during and after the infusion for S/S of a hypersensitivity reaction. In clinical trials, serious hypersensitivity reactions, including anaphylaxis, occurred during the infusion. S/S included chest tightness, dizziness, flushing, hypotension, nausea, urticaria, vomiting, and wheezing. Other hypersensitivity reactions occurred up to 3 hours after the start of the infusion and included angioedema, chest tightness, cough, erythema, flushing, nausea, pruritus, rash, throat irritation, and vomiting. Discontinue the infusion immediately if anaphylaxis occurs; consider the risk versus benefit of readministering taliglucerase in patients who have experienced a severe reaction. Caution should be exercised upon rechallenge. ■ Decreasing the infusion rate, temporarily stopping the infusion, and/or administering antihistamines, antipyretics, and/or corticosteroids is recommended. Pretreatment with antihistamines and/or corticosteroids may prevent subsequent reactions in these patients. ■ Observe patient closely for signs of improvement (e.g., improvement in hemoglobin and platelet counts, reduction in size of liver and/or spleen).
Patient education:
Enzyme replacement therapy is required for life. ■ Promptly report S/S of a hypersensitivity or infusion reaction (e.g., chills, dizziness, fatigue, fever, headache, itching, nausea, rash, shortness of breath, wheezing). Reactions may occur during or after infusion. ■ Pretreatment with antihistamines or corticosteroids may be used to prevent reactions.
Maternal/child:
Consider risk versus benefit. Women with Type 1 Gaucher disease have an increased risk of spontaneous abortion if disease symptoms are not treated and controlled before conception and during pregnancy. Pregnancy can exacerbate existing Type 1 Gaucher disease symptoms, and Type 1 Gaucher disease manifestations may lead to adverse pregnancy outcomes ■ Safety for use during breast-feeding not established. Not known if taliglucerase alfa is secreted in breast milk. The effects on the breast-fed infant or on milk production are also unknown. ■ Safety and effectiveness for use in pediatric patients under 4 years of age not established. Pediatric patients experienced a higher frequency of vomiting than adult patients. Frequencies of other adverse reactions were similar between adult and pediatric patients.
Elderly:
Numbers in clinical studies insufficient to determine if the elderly respond differently than do younger adults.
Drug/lab interactions
Specific information not available.
Side effects
The most commonly reported adverse reactions in clinical trials were abdominal pain, arthralgia, dizziness, extremity pain, fatigue, flushing, headache, nausea, pruritus, rash, urticaria, and vomiting. Back pain, diarrhea, and hypersensitivity reactions have also been reported.
Post-marketing:
Anaphylaxis.
Antidote
Keep physician informed of side effects; may be treated symptomatically if indicated. Based on the severity of the reaction, temporarily interrupt or discontinue infusion for clinical evidence of a hypersensitivity reaction. Patients have successfully continued therapy after mild hypersensitivity reactions with a reduction in rate of administration and/or treatment or pretreatment with antipyretics, antihistamines (e.g., diphenhydramine [Benadryl]), and/or corticosteroids (e.g., dexamethasone [Decadron]). Discontinue infusion immediately for anaphylaxis and treat with oxygen, epinephrine (Adrenalin), antihistamines (e.g., diphenhydramine [Benadryl]), vasopressors (e.g., dopamine), corticosteroids, albuterol, IV fluids, and ventilation equipment as indicated. Resuscitate as necessary.
Tedizolid phosphate
(te-DIZ-oh-lid FOS-fayt)
Sivextro
Antibacterial (oxazolidinone)
Usual dose
200 mg once daily for 6 days as an infusion over 1 hour. Alternately, may be given orally (with or without food).
Dose adjustments
No dose adjustment necessary when changing from IV to oral doses. ■ If a dose is missed or delayed, it should be administered as soon as possible any time up to 8 hours before the next scheduled dose. If less than 8 hours remain before the next dose, wait until the next scheduled dose. ■ No dose adjustments indicated based on age, gender, weight, race, or any degree of hepatic or renal impairment.
Dilution
Available in single-use vials containing 200 mg of tedizolid as a lyophilized powder. Reconstitute the 200-mg vial with 4 mL of SWFI. Gently swirl the contents and let the vial stand until completely dissolved and any foam disperses. Solution should be clear and colorless to pale yellow. Tilt the upright vial and insert a needle attached to a syringe to the bottom corner of the vial. Withdraw 4 mL of the reconstituted solution. Do not invert the vial during extraction of its contents. Further dilute by slowly injecting the 4 mL into a 250-mL bag of NS. Invert the bag gently to mix. Do NOT shake; may cause foaming.
Filters:
Specific information not available.
Storage:
Before use, vials may be stored at CRT. Total time from reconstitution to dilution to completion of administration of both reconstituted vials and/or fully diluted solutions should not exceed 24 hours at RT or under refrigeration at 2° to 8° C (36° to 46° F).
Compatibility
Tedizolid is compatible only with NS. Manufacturer states, “Other IV substances, additives, or other medications should not be added to tedizolid single-use vials or infused simultaneously through the same IV line or through a common IV port. If the same IV line is used for sequential infusion of additional medications, the line should be flushed before and after infusion of tedizolid with NS.” It is incompatible with any solution containing divalent cations (e.g., calcium, magnesium), including LR injection and Hartmann’s solution.
Rate of administration
A single dose as an infusion equally distributed over 1 hour. Do not administer as an IV push or bolus. If a common IV line is used to administer other drugs in addition to tedizolid, flush the IV line before and after each tedizolid infusion with NS.
Actions
Tedizolid phosphate belongs to the oxazolidinone class of antibacterial drugs. It is a phosphate prodrug and is converted to tedizolid by phosphatases. Its antibacterial activity is mediated by binding to the 50S subunit of the bacterial ribosome, resulting in inhibition of protein synthesis. This mechanism of action is different from that of other non–oxazodidinone class antibacterial drugs; therefore cross-resistance between tedizolid and other classes of antibacterial drugs is unlikely. Bacteriostatic against designated strains of Staphylococcus, Streptococcus, and Enterococcus; see Indications. Peak plasma concentrations are achieved at the end of the 1-hour infusion. Penetrates into the interstitial space fluid of adipose and skeletal muscle tissue with exposure similar to free-drug exposure in plasma. Approximately 70% to 90% bound to human plasma proteins. Half-life is approximately 12 hours. The majority of elimination occurs via the liver. Primarily excreted as a microbiologically inactive sulfate conjugate in feces with some excretion in urine.
Indications and uses
Treatment of adult patients with acute bacterial skin and skin structure infections (ABSSSI) caused by susceptible isolates of designated gram-positive microorganisms, including Staphylococcus aureus (both methicillin-susceptible [MSSA] and methicillin-resistant [MRSA] strains).
Contraindications
Manufacturer states, “None.”
Precautions
For IV use only. ■ Safety and efficacy for use in patients with neutropenia (neutrophil counts less than 1,000 cells/mm3) have not been adequately evaluated. In an animal model of infection, the antibacterial activity of tedizolid was reduced in the absence of granulocytes. Consider alternative therapy when treating patients with neutropenia and ABSSSI. ■ Specific sensitivity studies are indicated to determine susceptibility of the causative organism to tedizolid. ■ To reduce the development of drug-resistant bacteria and maintain its effectiveness, tedizolid should be used to treat only those infections proven or strongly suspected to be caused by bacteria. ■ Clostridium difficile–associated diarrhea (CDAD) has been reported for nearly all systemic antibacterial agents and may range in severity from mild diarrhea to fatal colitis. Consider in patients who present with diarrhea during or after treatment with tedizolid.
Monitor:
Obtain baseline CBC with differential and platelets. ■ Monitor for S/S of hypersensitivity (e.g., hypotension, rash, urticaria, tightness of the chest, wheezing).
Patient education:
Promptly report S/S of a hypersensitivity reaction (e.g., hives, rash, shortness of breath, wheezing). ■ Promptly report diarrhea or bloody stools that occur during treatment or up to several months after an antibiotic has been discontinued; may indicate CDAD and require treatment.
Maternal/child:
Category C: use during pregnancy only if the potential benefit outweighs the possible risk to the fetus. ■ Use caution during breast-feeding. ■ Safety and effectiveness for use in pediatric patients not established.
Elderly:
Numbers in clinical studies insufficient to determine if the elderly respond differently than do younger subjects. No overall differences in pharmacokinetics were observed between elderly subjects and younger subjects.
Drug/lab interactions
Tedizolid did not detectably inhibit or induce the metabolism of cytochrome P450 enzyme substrates. There was no degradation of tedizolid in human liver microsomes, indicating that tedizolid is unlikely to be a substrate for hepatic CYP450 enzymes. ■ During in vitro studies, no clinically significant inhibition of any transporter (e.g., the efflux transporter P-glycoprotein ([P-gp], selected OAT, OCT, or ABCG2) was observed. ■ A reversible inhibitor of monoamine oxidase. Not evaluated because subjects taking MAO inhibitors were excluded from trials. ■ In in vitro studies, tedizolid did not demonstrate synergy or antagonism with amphotericin B (conventional), aztreonam (Azactam), ceftazidime (Fortaz), ceftriaxone (Rocephin), ciprofloxacin (Cipro), clindamycin (Cleocin), daptomycin (Cubicin), gentamicin, imipenem-cilastin (Primaxin), ketoconazole (Nizoral), minocycline (Minocin), rifampin (Rifadin), terbinafine (Lamsil), sulfamethoxazole/trimethoprim (Bactrim), or vancomycin.
Side effects
The most common adverse reactions are diarrhea, dizziness, headache, nausea, and vomiting. CDAD, decreased WBC, dermatitis, eye disorders (e.g., asthenopia, blurred vision, visual impairment, vitreous floaters), flushing, hypersensitivity reactions, hypertension, hypoesthesia, increased hepatic transaminases, infusion-related reactions, insomnia, myelosuppression (anemia, neutropenia, thrombocytopenia), oral candidiasis, palpitations, paresthesia, pruritus, seventh nerve paralysis, tachycardia, urticaria, and vulvovaginal mycotic infection have been reported. Peripheral and optic neuropathy have been described in patients treated with another member of the oxazolidinone class (e.g., linezolid [Zyvox]) for longer than 28 days.
Antidote
Notify physician of any side effects. Discontinue the drug if indicated. Treat hypersensitivity reactions as indicated (e.g., diphenhydramine [Benadryl], epinephrine [Adrenalin], albuterol) and resuscitate as necessary. Mild cases of CDAD may respond to discontinuation of tedizolid. Treat CDAD with fluids, electrolytes, protein supplements, and oral vancomycin (Vancocin) or metronidazole (Flagyl) as indicated. In severe cases, surgical evaluation may be indicated. In the event of an overdose, discontinue tedizolid and provide supportive treatment as needed. Is not effectively removed from the circulation by hemodialysis.
Telavancin 
(tel-a-VAN-sin)
Vibativ
Antibacterial (lipoglycopeptide)
pH 4 to 5
Usual dose
Complicated skin and skin structure infections (cSSSI):
10 mg/kg as an infusion once every 24 hours for 7 to 14 days. Duration of therapy is dependent on severity and site of infection and on the patient’s clinical progress.
Hospital-acquired and ventilator-associated bacterial pneumonia (HABP/VABP):
10 mg/kg as an infusion once every 24 hours for 7 to 21 days. Duration of therapy is dependent on severity and site of infection and on the patient’s clinical progress.
Dose adjustments
Dose adjustment required in renal impairment as outlined in the following chart.
| Telavancin Dosage Adjustment in Adult Patients with Renal Impairment | |
| Creatinine Clearance* (mL/min) | Telavancin Dosage Regimen |
| >50 mL/min | 10 mg/kg every 24 hours |
| 30 to 50 mL/min | 7.5 mg/kg every 24 hours |
| 10 to <30 mL/min | 10 mg/kg every 48 hours |
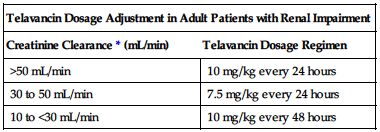
*Calculate using Cockcroft-Gault formula and ideal body weight. Use actual body weight if it is less than ideal body weight.
There is insufficient information to make specific dose recommendations for patients with end-stage renal disease (CrCl less than 10 mL/min), including patients undergoing hemodialysis. ■ Reduced doses may be indicated in the elderly based on age-related renal impairment. ■ No dose adjustment is recommended based on gender or in patients with mild or moderate hepatic impairment. Has not been studied in patients with severe hepatic impairment.
Dilution
Reconstitute each 250-mg vial with 15 mL of D5W, SWFI, or NS (45 mL for a 750-mg vial). Resultant solution has a final concentration of 15 mg/mL. To minimize foaming during reconstitution, allow the vacuum of the vial to pull the diluent from the syringe into the vial. Do not forcefully inject the diluent into the vial. Do not forcefully shake the vial. Reconstitution time is generally under 2 minutes but can occasionally take as long as 20 minutes. Mix thoroughly to dissolve contents completely. Discard vial if vacuum does not pull diluent into vial. Doses of 150 to 800 mg must be further diluted in 100 to 250 mL of D5W, NS, or LR. Doses less than 150 mg or greater than 800 mg must be diluted in a sufficient volume to provide a final concentration of 0.6 to 8 mg/mL. Do not shake the final infusion solution.
Storage:
Store unopened vials in refrigerator at 2° to 8° C (36° to 46° F). Excursions up to 25° C (77° F) are acceptable. Reconstituted and diluted solutions are stable for 12 hours at RT and for 7 days refrigerated. Total time in the vial plus the time in the infusion bag should not exceed 12 hours at RT and 7 days under refrigeration. The diluted solution can also be stored at −30° to −10° C (−22° to −14° F) for up to 32 days.
Compatibility (underline indicates conflicting compatibility information)
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
Manufacturer states, “Additives or other medications should not be added to telavancin single-use vials or infused simultaneously through the same IV line. If the same IV line is used for sequential infusion of additional medications, the line should be flushed before and after infusions of telavancin with D5W, NS, or LR.”
One source suggests the following compatibilities:
Y-site:
Not recommended by manufacturer. Amphotericin B lipid complex (Abelcet), ampicillin/sulbactam (Unasyn), azithromycin (Zithromax), calcium gluconate, caspofungin (Cancidas), cefepime (Maxipime), ceftazidime (Fortaz), ceftriaxone (Rocephin), ciprofloxacin (Cipro IV), colistimethate (Coly-Mycin M), cyclosporine (Sandimmune), dexamethasone (Decadron), diltiazem (Cardizem), dobutamine, dopamine, doripenem (Doribax), doxycycline, ertapenem (Invanz), famotidine (Pepcid IV), fluconazole (Diflucan), gentamicin, heparin, hydrocortisone sodium succinate (Solu-Cortef), imipenem-cilastatin (Primaxin), labetalol, magnesium sulfate, mannitol (Osmitrol), meropenem (Merrem IV), methylprednisolone (Solu-Medrol), metoclopramide (Reglan), milrinone (Primacor), norepinephrine (Levophed), ondansetron (Zofran), pantoprazole (Protonix IV), phenylephrine (Neo-Synephrine), piperacillin/tazobactam (Zosyn), potassium chloride (KCl), potassium phosphates, propofol (Diprivan), ranitidine (Zantac), sodium bicarbonate, sodium phosphates, tigecycline (Tygacil), tobramycin, vasopressin.
Rate of administration
A single dose equally distributed as an infusion over 60 minutes. Rapid IV infusions can cause “red man syndrome”–like infusion-related reactions, including flushing of the upper body, urticaria, pruritus, or rash. Stopping or slowing the infusion may result in cessation of these reactions.
Actions
A lipoglycopeptide antibacterial that is a synthetic derivative of vancomycin. Bactericidal against gram-positive organisms, including susceptible strains of staphylococci, streptococci, and vancomycin-susceptible enterococci. Exerts bactericidal action through inhibition of cell wall synthesis. Highly protein bound, primarily to albumin. The metabolic pathway for telavancin has not been identified. Half-life is 6.6 to 9.6 hours. Primarily excreted by the kidney.
Indications and uses
Treatment of adult patients with complicated skin and skin structure infections (cSSSI) caused by susceptible isolates of gram-positive microorganisms, including Staphylococcus aureus (including methicillin-susceptible and methicillin-resistant isolates), Streptococcus pyogenes, Streptococcus agalactiae, Streptococcus anginosus group, or Enterococcus faecalis (vancomycin-susceptible isolates only). ■ Treatment of adult patients with hospital-acquired and ventilator-associated bacterial pneumonia (HABP/VABP) caused by susceptible isolates of Staphylococcus aureus (including methicillin-susceptible and methicillin-resistant isolates). Reserve use for when alternative treatments are not suitable.
Contraindications
Known hypersensitivity to telavancin. ■ Use of IV unfractionated heparin sodium is contraindicated; see Drug/Lab Interactions.
Precautions
To reduce the development of drug-resistant bacteria and maintain its effectiveness, telavancin should be used to treat or prevent only those infections proven or strongly suspected to be caused by bacteria. ■ Sensitivity studies are necessary to determine susceptibility of the causative organism to telavancin. ■ Combination therapy may be clinically indicated if the documented or presumed pathogens include gram-negative organisms. ■ Prolonged use of drug may result in superinfection caused by overgrowth of nonsusceptible organisms. ■ New-onset or worsening renal impairment has been reported. Patients with underlying renal dysfunction or risk factors for renal dysfunction (diabetes mellitus, CHF, or hypertension) may be at increased risk. Patients who received concomitant medications known to affect kidney function (e.g., NSAIDs, ACE inhibitors, and loop diuretics) may also be at higher risk; see Drug Interactions. ■ Patients with pre-existing moderate to severe renal impairment (CrCl less than or equal to 50 mL/min) who were treated with telavancin for HABP/VABP had increased mortality observed versus vancomycin. Use of telavancin in patients with pre-existing moderate to severe renal impairment should be considered only when the anticipated benefit to the patient outweighs the potential risk. ■ Data from cSSSI trials suggest that clinical cure rates were lower in patients with baseline CrCl less than or equal to 50 mL/min. The same decrease in clinical cure rates was not seen in vancomycin-treated patients. These data should be considered when selecting antibacterial therapy for use in patients with baseline moderate/severe renal impairment. ■ Serious and sometimes fatal hypersensitivity reactions, including anaphylactic reactions, may occur after the first dose or subsequent doses. Telavancin is a semi-synthetic derivative of vancomycin; it is unknown if patients with hypersensitivity reactions to vancomycin will experience cross-reactivity to telavancin. Use with caution in patients with known hypersensitivity to vancomycin. ■ Clostridium difficile–associated diarrhea (CDAD) has been reported. May range from mild diarrhea to fatal colitis. Consider in patients who present with diarrhea during or after treatment with telavancin. ■ Infusion-related reactions have been reported; see Rate of Administration and Monitor. ■ Has caused prolongation of the QTc interval. Use with caution in patients who are taking drugs known to prolong the QTc interval. Avoid use in patients with congenital long QT syndrome, known prolongation of the QTc interval, uncompensated heart failure, or severe left ventricular hypertrophy. ■ Does not interfere with coagulation. Increased risk of bleeding and effects on platelet aggregation have not been observed. However, has been shown to affect certain anticoagulation tests; see Drug/Lab Interactions. ■ There is no known cross-resistance between telavancin and other classes of antibiotics. Some vancomycin-resistant enterococci have a reduced susceptibility to telavancin. ■ See Maternal/Child.
Monitor:
Obtain baseline CBC with differential and SCr. Monitor SCr every 48 to 72 hours, or more frequently if indicated, and at the end of therapy. If renal function deteriorates, the risk versus benefit of continuing therapy should be assessed. ■ Monitor for possible infusion-related reactions. ■ Monitor for S/S of hypersensitivity reactions.
Patient education:
Review manufacturer-supplied medication guide. ■ Women of childbearing potential should have a pregnancy test before initiating therapy. Effective birth control should be used throughout therapy. A pregnancy registry has been established to monitor the outcomes of women who become pregnant while receiving telavancin. ■ Report all side effects promptly. ■ Promptly report diarrhea or bloody stools that occur during treatment or up to several months after telavancin has been discontinued; may indicate CDAD and require treatment.
Maternal/child:
Category C: adverse developmental outcomes in three animal species at clinically relevant doses raise concerns about potential adverse developmental outcomes in humans. Avoid use during pregnancy unless potential benefit justifies potential risk. ■ Women with childbearing potential should have a serum pregnancy test before receiving telavancin. A pregnancy registry has been established to monitor pregnancy outcomes in women exposed to telavancin during pregnancy. ■ Safety for use in breast-feeding not established and effects unknown; use caution. ■ Safety and effectiveness for use in pediatric patients have not been studied.
Elderly:
In cSSSI trials, lower clinical cure rates were seen in patients over 65 years of age. Overall, treatment-emergent adverse events occurred with similar frequencies in patients of all age-groups studied. However, adverse events indicative of renal impairment occurred more frequently in the elderly. In HABP/VABP trials, treatment-emergent adverse events, deaths, and other serious adverse events occurred more often in patients 65 years of age or older. Consider age-related renal impairment; see Dose Adjustments.
Drug/lab interactions
Use with caution in patients taking medications known to prolong the QTc interval (e.g., amiodarone [Nexterone] and other antiarrhythmics, diphenhydramine [Benadryl], fosphenytoin [Cerebyx], furosemide [Lasix], itraconazole [Sporanox]). Effects may be additive. ■ Concomitant use with other agents that can affect renal function (e.g., NSAIDs [e.g., ibuprofen (Advil, Motrin), naproxen (Naprosyn, Aleve)], diuretics [e.g., furosemide (Lasix)], and ACE inhibitors [e.g., lisinopril (Zestril), enalapril (Vasotec)]) may increase the risk of renal toxicity. ■ Has been administered with aztreonam (Azactam) and piperacillin/tazobactam (Zosyn). There was no effect on the pharmacokinetics of either drug. ■ In vitro studies demonstrate no antagonism between telavancin and amikacin, aztreonam (Azactam), cefepime (Maxipime), ceftriaxone (Rocephin), ciprofloxacin (Cipro), gentamicin, imipenem-cilastatin (Primaxin), meropenem (Merrem), oxacillin (Bactocil), rifampin (Rifadin), and sulfamethoxazole/trimethoprim when tested against telavancin-susceptible staphylococci, streptococci, and enterococci. ■ May affect certain anticoagulation tests. Increases in PT, INR, aPTT, ACT, and coagulation-based factor X activity assays have been observed. Effects dissipate over time. Interference seen when using samples drawn between 0 and 18 hours after telavancin administration. Collect blood samples for these coagulation tests immediately before a patient’s next telavancin dose to minimize interaction. Because aPPT test results are expected to be artificially prolonged after televancin administration, concurrent use of intravenous unfractionated heparin is contraindicated. ■ Interferes with urine qualitative dipstick protein assays and quantitative dye methods (e.g., pyrogallol red-molybdate). However, microalbumin assays are not affected and can be used to monitor urinary protein excretion.
Side effects
The most common side effects include diarrhea, foamy urine, nausea, taste disturbances, and vomiting. Most serious side effects include cardiac events, CDAD, infusion-related reactions, nephrotoxicity (increased SCr, renal insufficiency, renal failure), and respiratory events. The most common events leading to discontinuation of therapy were acute renal failure, nausea, QTc prolongation, and rash. Other reported side effects include abdominal pain, decreased appetite, dizziness, infusion site pain and erythema, pruritus, and rigors.
Post-marketing:
Hypersensitivity reactions, including anaphylaxis.
Antidote
Notify the physician of all side effects. Initiate supportive care as indicated. Infusion-related reactions may respond to temporarily discontinuing or slowing the rate of infusion. Consider alternative antimicrobial therapy in patients who develop renal toxicity. Discontinue telavancin at the first sign of skin rash or other sign of hypersensitivity. Treat CDAD with fluids, electrolytes, protein supplements, and oral vancomycin (Vancocin) or metronidazole (Flagyl) as indicated. In severe cases, surgical evaluation may be indicated. There is no information on the use of hemodialysis or continuous venovenous hemofiltration in toxicity.
Temozolomide
(te-moe-ZOE-loe-mide)
Temodar
Antineoplastic (alkylating agent)
Usual dose
IV and oral doses are therapeutically equivalent if the IV dose is administered equally distributed over 90 minutes. The oral form should be used as soon as tolerated by the patient.
Newly diagnosed glioblastoma multiforme (GBM):
Concomitant phase:
75 mg/M2 daily for 42 days. Given concomitantly with focal radiotherapy. No dose reductions are recommended during the concomitant phase; however, dose interruptions or discontinuation may occur based on toxicity. The temozolomide dose should be continued throughout the 42-day concomitant period up to 49 days if all of the following conditions are met:
• Absolute neutrophil count must be equal to or greater than 1.5 × 109/L (1,500/mm3).
• Platelet count must be equal to or greater than 100 × 109/L (100,000/mm3).
• Nonhematologic toxicity must be equal to or less than Grade 1 (except for alopecia, nausea, and vomiting).
• CBC and platelet count should be obtained weekly.
• Interrupt or discontinue temozolomide dosing based on the hematologic and nonhematologic criteria in the following chart.
| Temozolomide (TMZ) Dosing Interruption or Discontinuation During Concomitant Radiotherapy | ||
| Toxicity | TMZ Interruption* | TMZ Discontinuation |
| Absolute neutrophil count | ≥0.5 and <1.5 × 109/L | <0.5 × 109/L |
| Platelet count | ≥10 and <100 × 109/L | <10 × 109/L |
| CTCAE nonhematologic toxicity (except for alopecia, nausea, vomiting) | CTCAE Grade 2 | CTCAE Grade 3 or 4 |
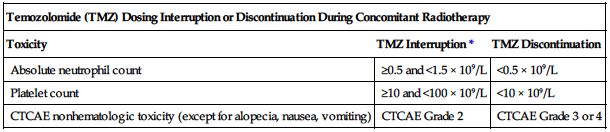
*Treatment with concomitant TMZ could be continued when all of the following conditions are met: ANC ≥1.5 × 109/L, platelet count ≥100 × 109/L, CTCAE nonhematologic toxicity Grade ≤1 (except for alopecia, nausea, vomiting).
TMZ, Temozolomide; CTCAE, Common Terminology Criteria for Adverse Events.
Prophylaxis against pneumocystis jiroveci pneumonia (PCP):
Required for all patients during this concomitant therapy. PCP prophylaxis should be continued in patients who develop lymphocytopenia until they recover from lymphocytopenia (CTCAE Grade ≤1).
Newly diagnosed glioblastoma multiforme (GBM):
Maintenance phase:
4 weeks after completing the temozolomide plus focal radiotherapy phase, temozolomide is administered for an additional 6 cycles of maintenance.
Cycle 1:
The initial dose is 150 mg/M2 once daily for 5 days followed by 23 days without treatment (a 28-day cycle).
Cycles 2 through 6:
The dose can be increased to 200 mg/M2 if the following conditions are met:
• Absolute neutrophil count is equal to or greater than 1.5 × 109/L (1,500/mm3)
• Platelet count is equal to or greater than 100 × 109/L (100,000/mm3)
• Nonhematologic toxicity is equal to or less than Grade 2 (except for alopecia, nausea, and vomiting).
The dose remains at 200 mg/M2 per day for the first 5 days of each subsequent cycle except if toxicity occurs. If the dose was not increased at Cycle 2, it should not be increased for subsequent cycles. See Dose Adjustments for dose reduction or discontinuation during maintenance.
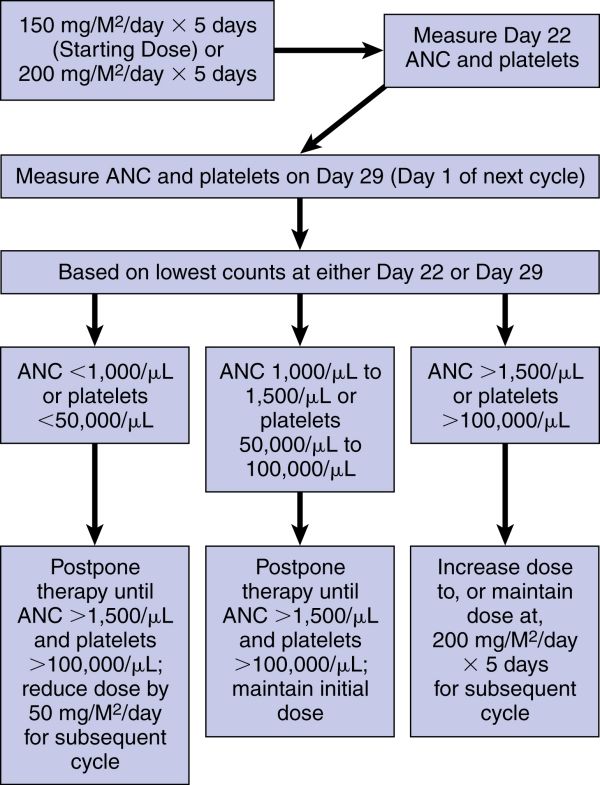
Refractory anaplastic astrocytoma:
150 mg/M2 once daily for 5 consecutive days for each 28-day treatment cycle. This dose may be increased to 200 mg/M2/day if both the nadir and day of dosing (Day 29, Day 1 of next cycle) ANC are equal to or greater than 1.5 × 109/L (1,500/mm3) and the nadir and day of dosing (Day 29, Day 1 of next cycle) platelet count are equal to or greater than 100 × 109/L (100,000/mm3). During treatment, a CBC should be obtained on Day 22 (21 days after the Day 1 dose of temozolomide) or within 48 hours of that day for each cycle. Repeat weekly until the ANC is above 1.5 × 109/L (1,500/mm3) and the platelet count exceeds 100 × 109/L (100,000/mm3). The next cycle of temozolomide should not be started until the ANC and platelet count exceed these levels. If the ANC falls to <1 × 109/L (1,000/mm3) or the platelet count is <50 × 109/L (50,000/mm3) during any cycle, reduce the next cycle by 50 mg/M2 but not below 100 mg/M2 (the lowest recommended dose). Therapy can be continued until disease progression. See the flow chart in Dose Adjustments.
Dose adjustments
Newly diagnosed glioblastoma multiforme (GBM):
Temozolomide dose must be adjusted according to the nadir ANC and platelet counts in the previous cycle and the ANC and platelet counts at the time of initiating the next cycle. Obtain a CBC and platelet count weekly during the concomitant phase and as indicated during the 4-week interim before beginning the maintenance phase. Obtain a baseline CBC and platelet count before beginning a cycle and repeat on Day 22 (21 days after the first dose of temozolomide in the cycle or within 48 hours of that day). Repeat weekly until the ANC is above 1.5 × 109/L (1,500/mm3) and the platelet count exceeds 100 × 109/L (100,000/mm3). The next cycle of temozolomide should not be started until the ANC and platelet count exceed these levels. This sequence is repeated for Cycles 1 through 6 of maintenance dosing. Base dose reductions during the next cycle on the lowest blood counts and worst nonhematologic toxicity during the previous cycle. Dose reductions or discontinuations during maintenance should be applied according to the following two charts.
| Temozolomide Dose Levels for Maintenance Treatment | ||
| Dose Level | Dose (mg/M2/day) | Remarks |
| −1 | 100 mg/M2/day | Reduction for prior toxicity |
| 0 | 150 mg/M2/day | Dose during Cycle 1 |
| 1 | 200 mg/M2/day | Dose during Cycles 2 through 6 in absence of toxicity |
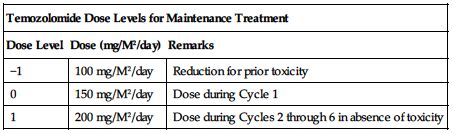
| Temozolomide Dose Reduction or Discontinuation During Maintenance Treatment | ||
| Toxicity | Reduce TMZ by 1 Dose Level* | Discontinue TMZ |
| ANC | <1 × 109/L | See † in footnote |
| Platelet count | <50 × 109/L | See † in footnote |
| CTCAE nonhematologic toxicity (except for alopecia, nausea, vomiting) | CTCAE Grade 3 | CTCAE Grade 4 |
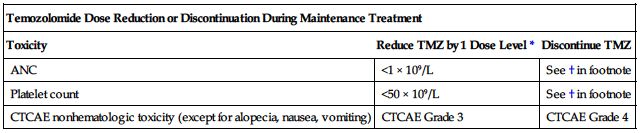
*See preceding chart for temozolomide dose levels for maintenance treatment.
†TMZ is to be discontinued if a dose reduction to <100 mg/M2 is required or if the same Grade 3 nonhematologic toxicity (except for alopecia, nausea, vomiting) recurs after dose reduction.
TMZ, Temozolomide; CTCAE, Common Terminology Criteria for Adverse Events.
See the next page for temozolomide dose modifications for refractory anaplastic astrocytoma.
Temozolomide Dose Modification Table for Refractory Anaplastic Astrocytoma:
Dilution
Specific techniques required; see precautions.
Bring vial(s) to room temperature before reconstitution. Reconstitute each 100-mg vial with 41 mL of SWFI. Swirl gently to dissolve; do not shake; will yield a concentration of 2.5 mg/mL of temozolomide. Do not further dilute the reconstituted solution. Withdraw up to 40 mL from each vial and transfer into an empty 250-mL PVC infusion bag for delivery with an infusion pump.
Filters:
Specific information not available.
Storage:
Refrigerate single-use vials at 2° to 8° C (36° to 46° F). Store reconstituted product at RT. Reconstituted solution must be used within 14 hours, including infusion time.
Compatibility
No compatibility data available. Manufacturer states, “Other medications should not be infused simultaneously through the same IV line.” May be administered only in the same infusion line as NS.
Rate of administration
A single dose equally distributed over 90 minutes. Use of an infusion pump is recommended. Bioequivalence was established only with the 90-minute infusion. Infusion over a longer or shorter period may result in suboptimal dosing. In addition, the possibility of an increase in infusion-related adverse reactions cannot be ruled out. Flush IV lines before and after temozolomide infusion. Infusion must be complete within 14 hours of reconstitution.
Actions
An imidazotetrazine derivative and alkylating agent. It is not directly active but undergoes rapid nonenzymatic conversion, as dacarbazine does, to the reactive compound 5-(3-methyl triazen-1-yl)-imidazole-4-carboxamide (MTIC) and to temozolomide acid metabolite. Cytotoxicity is thought to be due primarily to alkylation of DNA. Weakly bound to plasma proteins. Rapidly eliminated with a mean elimination half-life of 1.8 hours. Some excretion in urine and a very small amount in feces.
Indications and uses
Treatment of adults with newly diagnosed glioblastoma multiforme. Initially given concomitantly with radiotherapy and then continued as maintenance treatment. ■ Treatment of adults with refractory anaplastic astrocytoma (i.e., adults who have experienced disease progression on a drug regimen containing nitrosourea and procarbazine).
Contraindications
Known history of hypersensitivity reaction to temozolomide or any of its components (e.g., urticaria, allergic reaction including anaphylaxis, toxic epidermal necrolysis, and Stevens-Johnson syndrome). ■ Known history of hypersensitivity to dacarbazine.
Precautions
Follow guidelines for handling cytotoxic agents. See Appendix A, p. 1331. ■ Usually administered by or under the direction of the physician specialist in a facility with adequate diagnostic and treatment facilities to monitor the patient and respond to any medical emergency. ■ Myelosuppression may be severe and dose limiting. May include prolonged pancytopenia, which may result in aplastic anemia. Deaths have been reported. Risk may be increased with concomitant use of other medications associated with aplastic anemia (e.g., carbamazepine [Tegretol], phenytoin [Dilantin], sulfamethoxazole/trimethoprim [Bactrim]). ■ Risk of myelosuppression increased in women and the elderly. ■ Cases of myelodysplastic syndrome and secondary malignancies, including myeloid leukemia, have been observed. ■ Prophylaxis against Pneumocystis jiroveci pneumonia (PCP) is required for all patients being treated for newly diagnosed glioblastoma multiforme (a 42-day regimen). The longer dosing regimen may increase the risk of PCP; however, all patients, particularly those receiving steroids, should be monitored for symptoms of PCP regardless of the regimen. ■ Severe, sometimes fatal hepatotoxicity have been reported. ■ Use caution in patients with severe renal or hepatic impairment.
Monitor:
Obtain a baseline CBC and platelet count. See Usual Dose for minimum levels required before administration of temozolomide. ■ Obtain baseline liver function tests. Repeat midway through the first cycle, before each subsequent cycle, and approximately 2 to 4 weeks after the last dose of temozolomide.
Patients with newly diagnosed GBM:
Obtain a CBC and platelet count weekly during the concomitant phase and as indicated during the 4-week interim before beginning the maintenance phase. Obtain a baseline CBC and platelet count before beginning a maintenance dose (28-day cycles) and repeat on Day 22 (21 days after the first maintenance dose of temozolomide or within 48 hours of that day). Repeat weekly until the ANC is above 1.5 × 109/L (1,500/mm3) and the platelet count exceeds 100 × 109/L (100,000/mm3). This sequence is repeated for Cycles 1 through 6 of maintenance dosing.
Patients with refractory anaplastic astrocytoma:
Obtain a CBC and platelet count on Day 1 of each cycle. During treatment (28-day cycles), a CBC should be obtained on Day 22 (21 days after the Day 1 dose of temozolomide or within 48 hours of that day for each cycle). Repeat weekly until the ANC is above 1.5 × 109/L (1,500/mm3) and the platelet count exceeds 100 × 109/L (100,000/mm3). Myelosuppression usually occurred within the first few cycles of therapy and was not cumulative. It occurred late in the treatment cycle and returned to normal, on average, within 14 days of nadir counts. (The median nadirs occurred at 26 days for platelets and 28 days for neutrophils.)
All indications:
Nausea and vomiting may be significant. Prophylactic antiemetics may reduce nausea and vomiting and increase patient comfort. ■ Observe closely for signs of infection. Prophylactic antibiotics may be indicated pending results of C/S in a febrile neutropenic patient. ■ Monitor for thrombocytopenia (platelet count less than 50,000/mm3). Initiate precautions to prevent excessive bleeding (e.g., inspect IV sites, skin, and mucous membranes; use extreme care during invasive procedures; test urine, emesis, stool, and secretions for occult blood). ■ Regardless of the regimen, monitor all patients, particularly those receiving steroids, for symptoms of PCP (dyspnea; fever; dry, nonproductive cough; characteristic x-ray).
Patient education:
Avoid pregnancy; use of nonhormonal birth control is recommended. ■ Promptly report a rash; swelling of the face, throat, or tongue; severe skin reaction; or troubled breathing. ■ Report IV site burning or stinging promptly. ■ Secondary malignancies have been reported. ■ See Appendix D, p. 1333. ■ Additional precautions are required with the capsule form.
Maternal/child:
Category D: avoid pregnancy; can cause fetal harm. ■ Discontinue breast-feeding. ■ Safety and effectiveness for use in pediatric patients not established. A 5-day regimen every 28 days using the oral formulation has been studied in selected pediatric patients from 3 to 18 years of age. Toxicity profile was similar to that seen in adults.
Elderly:
Numbers in clinical studies are insufficient to determine if the elderly respond differently than younger subjects. Dose selection should be cautious based on the potential for decreased organ function and concomitant disease or drug therapy. In newly diagnosed patients with glioblastoma multiforme, side effects were similar to those seen in younger patients. In the anaplastic astrocytoma study, patients 70 years or older had a higher incidence of Grade 4 neutropenia and Grade 4 thrombocytopenia in the first cycle of therapy.
Drug/lab interactions
Valproic acid decreases the oral clearance of temozolomide by about 5%; clinical significance is not known. ■ Administration of live attenuated viral or bacterial vaccines should be avoided.
Side effects
Newly diagnosed glioblastoma multiforme:
Myelosuppression may be severe and dose limiting. May include prolonged pancytopenia, which may result in aplastic anemia; deaths have been reported. Alopecia, anorexia, constipation, headache, nausea and vomiting, and thrombocytopenia were the most frequently reported side effects. The most commonly reported severe or life-threatening reactions were convulsions, fatigue, headache, and thrombocytopenia. Abdominal pain, arthralgia, blurred vision, confusion, coughing, diarrhea, dizziness, dry skin, dyspnea, erythema, hypersensitivity reactions (including anaphylaxis), insomnia, memory impairment, pruritus, radiation injury, rash, stomatitis, taste perversion, and weakness have also been reported.
Refractory anaplastic astrocytoma:
Fatigue, headache, and nausea and vomiting were the most frequently reported side effects. Myelosuppression (neutropenia and thrombocytopenia) was the dose-limiting adverse reaction. Abdominal pain, abnormal coordination, abnormal gait, abnormal vision (blurred vision, vision changes, visual deficit), adrenal hypercorticism, amnesia, anemia, anorexia, anxiety, asthenia, ataxia, back pain, breast pain (female), confusion, constipation, convulsions (local and general), coughing, depression, diarrhea, diplopia, dizziness, dysphasia, fever, hemiparesis, insomnia, lymphopenia, myalgia, paresis, paresthesia, peripheral edema, pharyngitis, pruritus, rash, sinusitis, somnolence, URI, urinary incontinence, UTI, viral infection, and weight increase have been reported.
Injection site reactions:
Erythema, irritation, pain, pruritus, swelling, and warmth at the infusion site; hematoma; and petechiae.
Post-marketing:
Alveolitis, cholestasis, diabetes insipidus, elevation of liver enzymes, erythema multiforme, hepatitis, hepatotoxicity (severe and sometimes fatal), hyperbilirubinemia, hypersensitivity reactions (including anaphylaxis), opportunistic infections (including pneumocystis pneumonia [PCP], primary and reactivated cytomegalovirus [CMV], and reactivation of hepatitis B), interstitial pneumonitis, pneumonitis, prolonged pancytopenia that may result in aplastic anemia with fatal outcomes, pulmonary fibrosis, Stevens-Johnson syndrome, and toxic epidermal necrolysis.
Antidote
Keep physician informed of all side effects and CBC results. Temozolomide may need to be reduced or discontinued. Symptomatic and supportive treatment is indicated. Administration of whole blood products (e.g., packed RBCs, platelets, leukocytes) and/or blood modifiers (e.g., darbepoetin alfa [Aranesp], epoetin alfa [Epogen], filgrastim [Neupogen, Zarxio], pegfilgrastim [Neulasta], sargramostim [Leukine]) may be indicated to treat bone marrow toxicity. Precautions are indicated for cancer patients with erythropoietin-stimulating agents (ESAs); see darbepoetin alfa and epoetin alfa monographs. Treat hypersensitivity reactions and/or anaphylaxis as indicated (e.g., epinephrine, corticosteroids, oxygen, and antihistamines [diphenhydramine]). There is no specific antidote. Resuscitate as indicated.
Temsirolimus
(TEM-sir-OH-li-mus)
Torisel
Antineoplastic (kinase inhibitor)
Usual dose
Premedication:
To minimize the incidence of hypersensitivity reactions, administer diphenhydramine (Benadryl) 25 to 50 mg IV (or similar antihistamine) 30 minutes before the start of each dose of temsirolimus; see Precautions and Monitor.
Temsirolimus:
25 mg as an infusion once a week until disease progression or unacceptable toxicity.
Dose adjustments
Hold for absolute neutrophil count (ANC) less than 1,000/mm3, platelet count less than 75,000/mm3, or CTCAE Grade 3 or greater adverse reactions. Once toxicities have resolved to Grade 2 or less, restart therapy with the dose reduced by 5 mg/week to a dose no lower than 15 mg/week. ■ Consider dose reduction to 12.5 mg/week when coadministered with a strong CYP3A4 inhibitor; see Drug Interactions. If the strong inhibitor is discontinued, wait approximately 1 week before adjusting the temsirolimus dose upward to the indicated dose. ■ Consider a dose increase to 50 mg/week when coadministered with a strong CYP3A4 inducer; see Drug Interactions. If the strong inducer is discontinued, the temsirolimus dose should be returned to the dose used prior to initiation of the strong CYP3A4 inducer. ■ Decrease dose to 15 mg/week in patients with mild hepatic impairment (bilirubin greater than 1 to 1.5 ULN or AST greater than ULN but bilirubin equal to or less than ULN); see Contraindications. ■ No dose adjustment indicated based on age, race, gender, or renal status. Not studied in hemodialysis patients.
Dilution
Specific techniques required; see precautions and compatibility.
Available as a kit containing a vial of temsirolimus and a vial of a manufacturer-supplied diluent. Temsirolimus and diluent vials contain overfill. A two-step dilution process is required. Reconstitute the temsirolimus vial with 1.8 mL of the provided diluent. Invert the vial several times to mix thoroughly. Allow time for foam to subside. Final concentration in temsirolimus vial is 10 mg/mL. Solution will be clear to slightly turbid and colorless to yellow. Withdraw the required amount of reconstituted temsirolimus and inject into a 250-mL container (glass, polyolefin, or polypropylene) of NS. Invert bag to mix; see Compatibility. Avoid excessive shaking. Use of non-DEHP, non-polyvinylchloride (PVC) tubing (polyethylene-lined administration set is recommended) with appropriate filter is recommended. If a PVC administration set must be used, it should not contain DEHP. Temsirolimus contains polysorbate 80 when diluted, which increases the rate of DEHP extraction from PVC.
Filter:
Use of an in-line polyethersulfone filter with a pore size not greater than 5 microns is recommended. If an administration set with an in-line filter is not available, a polyethersulfone end-filter should be used. The use of both an in-line and end-filter is not recommended.
Storage:
Store unopened vials at 2° to 8° C (36° to 46° F). Protect from light. During handling and preparation, protect from excessive room light or sunlight. The temsirolimus/diluent mixture (10 mg/mL) is stable for up to 24 hours below 25° C (77° F) in the temsirolimus vial. Administration of the final product diluted in NS should be completed within 6 hours of adding the temsirolimus/diluent mixture to the NS.
Compatibility
Manufacturer states, “Final dilution for infusion should be stored in bottles (glass, polypropylene) or plastic bags (polypropylene, polyolefin) and administered through polyethylene-lined administration sets.” Non–DEHP-containing materials must be used for administration. Manufacturer also states that undiluted temsirolimus should not be added directly to aqueous infusion solutions. A precipitate will form. In addition, “The stability of temsirolimus in other infusion solutions has not been evaluated. Addition of other drugs or nutritional agents to admixtures of temsirolimus in NS has not been evaluated and should be avoided. Temsirolimus is degraded by both acids and bases, and thus combinations of temsirolimus with agents capable of modifying solution pH should be avoided.”
Rate of administration
A single dose as an infusion equally distributed over 30 to 60 minutes. Use of an infusion pump is recommended. Increase duration of infusion to 60 minutes in patients who experience a hypersensitivity reaction.
Actions
An inhibitor of mTOR (mammalian target of rapamycin). Binds to an intracellular protein (FKBP-12); this protein-drug complex inhibits the activity of mTOR that controls cell division. Results in a cell cycle–specific (G1) growth arrest in treated tumor cells. In in vitro studies, temsirolimus inhibited the activity of mTOR and resulted in reduced levels of hypoxia-inducible factors HIF-1α and HIF-2α and the vascular endothelial growth factor. Both temsirolimus and sirolimus are extensively partitioned into formed blood elements. Extensively metabolized, primarily by cytochrome P450 3A4. Sirolimus is the principal metabolite and is active. Elimination is primarily via the feces and to a small extent via urine. Mean half-lives of temsirolimus and sirolimus were 17.3 and 54.6 hours, respectively.
Indications and uses
Treatment of advanced renal cell carcinoma.
Contraindications
Bilirubin greater than 1.5 ULN; see Dose Adjustments and Precautions.
Precautions
Follow guidelines for handling cytotoxic agents. See Appendix A, p. 1331. ■ Should be administered by or under the direction of the physician specialist in facilities equipped to monitor the patient and respond to any medical emergency. ■ Hypersensitivity/infusion reactions have been reported. May occur early in the first infusion but may also occur with subsequent infusions. Premedication required to minimize the chance of a reaction; see Usual Dose. Use with caution in patients with known hypersensitivity to temsirolimus or its metabolites (including sirolimus) or to any components of the product. Use caution in patients who cannot receive prophylactic treatment with an antihistamine for medical reasons; see Monitor. ■ Patients with moderate and severe hepatic impairment had increased rates of adverse reactions and deaths. Use caution when treating patients with mild hepatic impairment; see Dose Adjustments and Contraindications. ■ Hyperglycemia/glucose intolerance is common. Initiation of or an increase in the dose of insulin and/or oral hypoglycemic agent therapy may be required. ■ Elevated cholesterol and triglycerides are common. Initiation or adjustment of existing therapy may be required. ■ May cause immunosuppression, thus increasing the risk of infections, including opportunistic infections. Pneumocystis jiroveci pneumonia (PJP), including fatalities, has been reported. May be associated with concomitant use of corticosteroids or other immunosuppressants. ■ Cases of interstitial lung disease, some resulting in death, have been reported. ■ Cases of fatal bowel perforation have been reported. ■ Rapidly progressive and sometimes fatal acute renal failure has occurred. ■ Has been associated with abnormal wound healing. Exercise caution when temsirolimus is used in the perioperative period. ■ Patients who have CNS tumors and/or are receiving anticoagulation therapy may be at increased risk of intracerebral bleeding. ■ The use of live virus vaccines and close contact with those who have received live virus vaccines should be avoided. See Drug Interactions.
Monitor:
Obtain baseline and weekly CBC with differential and platelet count. Obtain a baseline chemistry panel, including bilirubin and liver function tests (ALT, AST), and repeat every 2 weeks. Obtain baseline radiographic assessment by lung CT or chest x-ray. Repeat assessments periodically. ■ Monitor for S/S of a hypersensitivity/infusion reaction (e.g., anaphylaxis, apnea, bronchospasm, chest pain, dyspnea, flushing, hypotension, loss of consciousness, rash). If a reaction occurs, the infusion should be stopped and the patient should be observed for at least 30 to 60 minutes (depending on the severity of the reaction). At the discretion of the physician, and after a benefit-risk assessment, treatment may be resumed with the administration of an H1-receptor antagonist (such as diphenhydramine [Benadryl]) if not previously administered and/or an H2-receptor antagonist (e.g., ranitidine [Zantac] 50 mg IV or famotidine [Pepcid] 20 mg IV) approximately 30 minutes before restarting the temsirolimus infusion. The infusion may be resumed at a slower rate; see Rate of Administration. ■ Monitor serum glucose, cholesterol, and triglycerides before and during therapy. ■ Monitor for S/S of infection. ■ Consider prophylaxis of PJP when concomitant use of corticosteroids or other immunosuppressive agents are required. ■ Monitor respiratory status. Patients may present with symptoms such as dyspnea, cough, hypoxia, and fever or may be asymptomatic. If clinically significant respiratory symptoms develop, consider withholding temsirolimus until after recovery of symptoms and improvement of radiographic findings related to pneumonitis. Opportunistic infections such as PJP should be considered in the differential diagnosis. Empiric treatment with corticosteroids and/or antibiotics may be considered. ■ Monitor for S/S of bowel perforation (abdominal pain, acute abdomen, bloody stools, diarrhea, fever, metabolic acidosis). ■ Monitor for thrombocytopenia (platelet count less than 50,000/mm3); see Dose Adjustments and Contraindications. Initiate precautions to prevent excessive bleeding (e.g., inspect IV sites, skin, and mucous membranes; use extreme care during invasive procedures; test urine, emesis, stool, and secretions for occult blood).
Patient education:
Review medical history with health care provider. Temsirolimus may affect existing medical conditions (e.g., diabetes, high cholesterol, hyperlipidemia). Therapy may need to be adjusted. ■ Review medication list (prescription, over-the-counter, and herbal [including past or present use of corticosteroids or immunosuppressants]) with health care provider. Interactions are possible. ■ Promptly report any S/S of a hypersensitivity reaction (chest tightness, dyspnea, flushing, pruritus, rash, urticaria). ■ Avoid pregnancy during and for 3 months after completion of therapy. Men with partners of childbearing potential should use reliable contraception during treatment and for 3 months after completion of therapy. ■ Report excessive thirst or any increase in volume or frequency of urination. ■ Promptly report S/S of infection, new or worsening respiratory symptoms, new or worsening abdominal pain, or blood in stools. ■ Wound healing complications may occur while on therapy. ■ The use of live virus vaccines and close contact with those who have received live virus vaccines should be avoided. ■ Increased risk of intracerebral bleed in patients who have CNS tumors and/or are receiving anticoagulants.
Maternal/child:
Category D: avoid pregnancy; may cause fetal harm. ■ Potential for tumorigenicity. Discontinue breast-feeding. ■ Safety and effectiveness for use in pediatric patients not established.
Elderly:
Elderly patients may be more likely to experience certain side effects, including diarrhea, edema, and pneumonia.
Drug/lab interactions
Concomitant use of strong CYP3A4 inhibitors (e.g., amprenavir [Agenerase], atazanavir [Reyataz], clarithromycin [Biaxin], delavirdine [Rescriptor], indinavir [Crixivan], itraconazole [Sporanox], ketoconazole [Nizoral], nefazodone, nelfinavir [Viracept], ritonavir [Norvir], saquinavir [Invirase], telithromycin [Ketek], or voriconazole [VFEND]) may increase plasma concentrations of the active metabolite sirolimus. Avoid concomitant use if possible. If required, consider a dose reduction of temsirolimus to 12.5 mg/week and monitor closely for acute toxicity. If the strong inhibitor is discontinued, a washout period of approximately 1 week should be allowed before adjusting the temsirolimus dose upward to the indicated dose; see Dose Adjustments. ■ Grapefruit juice may increase plasma concentrations of sirolimus (a major metabolite of temsirolimus) and should be avoided. ■ CYP3A4/5 inducers (e.g., carbamazepine [Tegretol], dexamethasone [Decadron], phenobarbital [Luminal], phenytoin [Dilantin], rifabutin [Mycobutin], rifampin [Rifadin]) may decrease plasma concentrations of the active metabolite sirolimus, thus decreasing effectiveness. Avoid concomitant use if possible. If required, consider a dose increase to 50 mg/week. If the strong inducer is discontinued, the temsirolimus dose should be returned to the dose used before initiating the strong CYP3A4 inducer; see Dose Adjustments. ■ Angioneurotic edema-type reactions (including delayed reactions occurring 2 months after initiation of therapy) have been observed in some patients with concurrent administration of ACE inhibitors (e.g., enalapril [Vasotec]). ■ St. John’s wort may decrease temsirolimus plasma concentrations, decreasing effectiveness. Avoid use. ■ Use caution with other drugs that inhibit the efflux transporter P-glycoprotein (P-gp, ABCB1) (e.g., protease inhibitors [e.g., nelfinavir (Viracept)]); may increase concentrations of temsirolimus. ■ Temsirolimus inhibits P-gp in vitro. Coadministration with drugs that are substrates of P-gp may result in increased concentration of substrates. ■ The combination of sunitinib (Sutent) and temsirolimus resulted in dose-limiting toxicity. ■ Do not administer live virus vaccines to patients receiving temsirolimus, and avoid close contact with those who have received live virus vaccines. ■ No clinically significant effect is anticipated when temsirolimus is coadministered with agents that are metabolized by CYP2D6 or CYP3A4.
Side effects
The most common side effects include anemia, anorexia, asthenia, edema, elevated alkaline phosphatase and AST, elevated SCr, hyperglycemia, hyperlipemia, hypertriglyceridemia, hypophosphatemia, leukopenia, lymphopenia, mucositis, nausea, rash, and thrombocytopenia. Other commonly reported side effects include abdominal pain, acne, arthralgia, chest pain, chills, constipation, convulsions, cough, depression, diarrhea, dry skin, dysgeusia, dyspnea, edema, epistaxis, fever, headache, hemorrhage (GI, rectal), hypokalemia, increased bilirubin, increased total cholesterol, increased triglycerides, infection (pneumonia, URI, wound [including postoperative] sepsis), insomnia, nail disorder, neutropenia, pain, pericardial effusion, pleural effusion, pneumonitis, pruritus, rash, vomiting, and weight loss. The most serious side effects include bowel perforation, hyperglycemia/glucose intolerance, hyperlipemia, hypersensitivity reactions, hypertension, interstitial lung disease, intracerebral hemorrhage, renal failure, venous thromboembolism (including DVT and pulmonary embolus [including fatal outcomes]), and wound healing complications.
Post-marketing:
Complex regional pain syndrome (reflex sympathetic dystrophy); extravasation with erythema, pain, swelling, and warmth; injection site reactions; rhabdomyolysis; Stevens-Johnson syndrome.
Antidote
Keep physician informed of all side effects. Most minor side effects will be treated symptomatically. Monitor patient closely. Discontinue temsirolimus at the first sign of a hypersensitivity reaction. Monitor patient for 30 to 60 minutes. Treatment may be resumed at the discretion of the physician; see Monitor. Treat severe hypersensitivity reactions as indicated; may require epinephrine, airway management, oxygen, IV fluids, antihistamines (e.g., diphenhydramine [Benadryl]), corticosteroids (e.g., hydrocortisone sodium succinate [Solu-Cortef]), and pressor amines (e.g., dopamine). Treat the development of interstitial lung disease as indicated. May require discontinuation of temsirolimus and/or treatment with corticosteroids and/or antibiotics. Some patients have been able to continue therapy without additional intervention.
Tenecteplase
(teh-NECK-teh-plays)
TNKase
Thrombolytic agent (recombinant)
pH 7.3
Usual dose
Initiate therapy as soon as possible after the onset of acute myocardial infarction (AMI) symptoms. Total dose is based on patient weight and should not exceed 50 mg. See the following chart.
| Tenecteplase Dosing Guidelines | ||
| Patient Weight (kg) | Tenecteplase (mg) | Volume of Tenecteplase* to Be Administered (mL) |
| <60 kg | 30 mg | 6 mL |
| ≥60 to <70 kg | 35 mg | 7 mL |
| ≥70 to <80 kg | 40 mg | 8 mL |
| ≥80 to <90 kg | 45 mg | 9 mL |
| ≥90 kg | 50 mg | 10 mL |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


