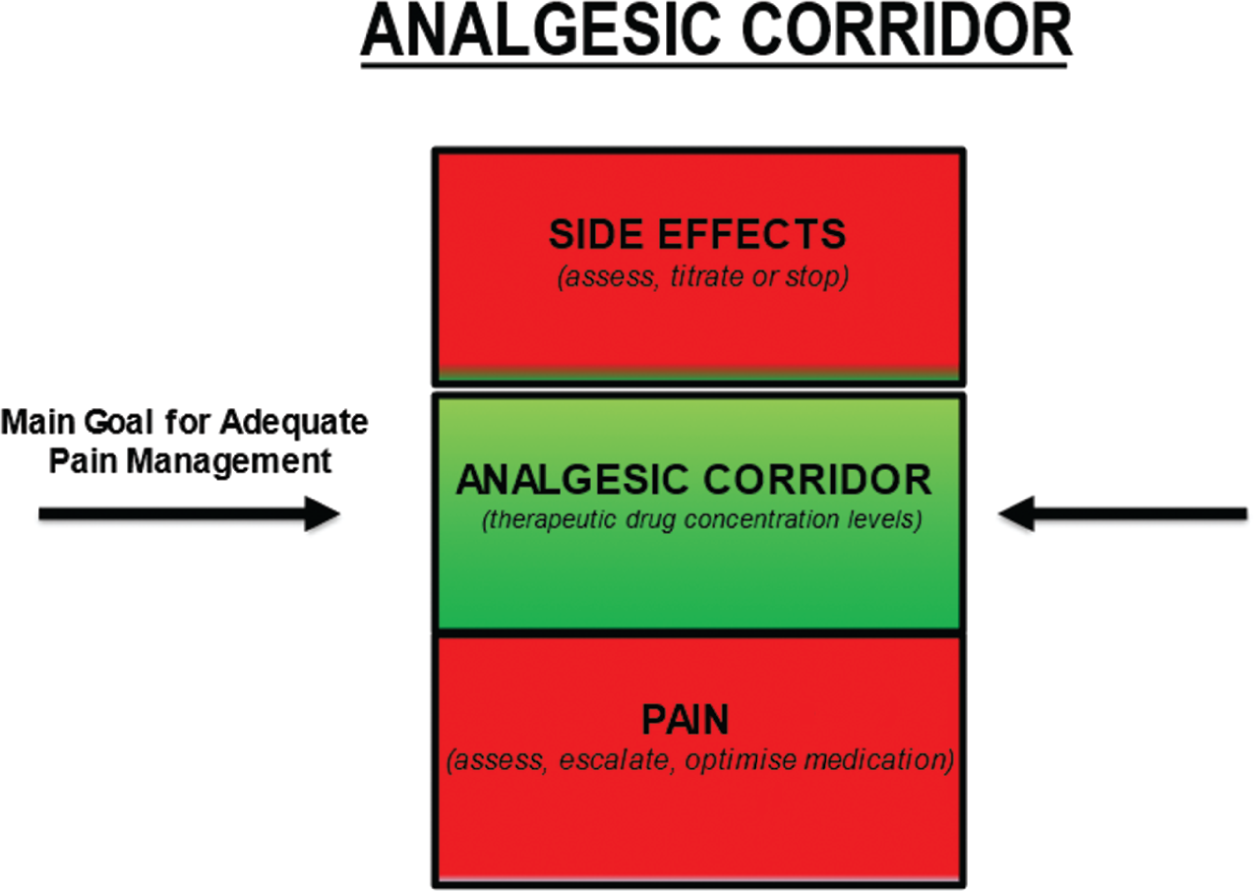
Debbie Omodele, Danielle Edge and Doreen Crawford This chapter will explore the care of a patient with sickle cell disease (SCD), which is the most common genetic anomaly in the UK today, affecting over 1 in every 2000 live births (De et al. 2019; SCD standards, 2019). It is an autosomal recessive disorder and causes very significant morbidity and mortality. SCD predominantly affects people with African, Caribbean, and Mediterranean ancestry, however, with the ever-growing diversity of the population and inter-racial families, it can affect people of any race and ethnicity. SCD results from the presence of a mutated form of haemoglobin, haemoglobin S (HbS). The main symptoms of sickle cell disease are anaemia and episodes of severe pain, but there are a number of other complications such as stroke, acute chest syndrome, priapism, and jaundice. SCD is a chronic long term life-limiting condition, knowledge of SCD is therefore important for the children’s nurse as the disease has serious implications for the quality of life (QoL) and the life chances of children (Baker et al. 2021). A comprehensive review of genetics is beyond the scope of this chapter however SCD is known as an autosomal recessive disorder which means that both copies of an affected gene must be present to cause the condition (Davies & Meimaridou 2020). People with only one defective allele in the gene are known as carriers of the sickle gene. When both parents are carriers, there is a 25% chance that a child will inherit two copies of the gene (one from each parent) and develop disease. The chance of the child inheriting one normal and one abnormal copy of the gene, so they are also a carrier of the disease, is 50%, and the chance of them inheriting no affected copy of the gene is 25%. In both these instances, the child will not inherit the condition. Each pregnancy will have a 1 in 4 chance of the offspring having the condition (Davies & Meimaridou 2020). All pregnant women are offered screening for SCD and other unusual haemoglobins and therefore make an informed decision to either accept or decline screening. The parents of all newborn infants born in the UK are offered screening for SCD as part of the Newborn Blood Spot Programme (2018), which identifies conditions that can affect a child’s long-term health or survival. This is important because early management greatly reduces the serious symptoms the disease can cause. Newborn screening is important as a method of early detection which triggers prompt referral into the specialist care pathway and timely follow-up allowing infants to be seen at a paediatric clinic by three months of age (Sickle Cell Disease in Childhood 2019). This allows for prophylactic treatment with antibiotics (penicillin/erythromycin) to be offered and initiated by three months of age which has shown to improve quality of life by reducing the risk of infections (Cober & Phelps 2010). Diagnosis could also be made antenatally via prenatal diagnosis (PND), which is offered to couples at risk of having a baby with a significant haemoglobinopathy or via a simple venepuncture blood test for those who have not undergone screening for whatever reason. The hallmarks of SCD include episodes of severe pain. Pain in SCD is unpredictable and often recurrent episodes due to vaso-occulsive crises, with CYP experiencing more acute episodes than chronic pain seen in the adult population (Inusa 2016). Therefore, it is essential for children’s nurses and HCP’s to understand the principles of achieving effective pain management by targeting and keeping patients within the therapeutic window of prescribed analgesia, maximising its effects. Ongoing management of CYP with SCD includes regular follow-up hospital appointments, encouraging adherence to treatment, immunisations including additional pneumococcal vaccinations from the age of 2 years old, while supporting, educating, and empowering families to become experts with managing their child’s care. Other long-term preventative measures to improve quality of life include regular blood transfusion therapy for stroke prevention and hydroxycarbamide for managing frequency and severity of a sickle cell crisis (Sickle Cell Disease in Childhood 2019). Listening to the CYP and families voice about their ongoing experience is essential to long term management of SCD. Foster and Ellis (2018) systematic review highlighted the need for support for the CYP that is collaborative across health, education, and families. Refer to Box Activity 37.2. This introduction to SCD has considered what the disease is, the genetics and population screening for SCD, and treatment and management of the long-term condition. The content within this chapter will explore the case study and focus on aspects of care planning needs and questions to consider when caring for a child presenting in a SCD crisis. Finally, we will take a look to the future and consider what new treatment research might provide and further education healthcare professionals need to deliver CYP’s the best evidence-based care. Box Activity 37.3. Throughout this section the nursing process has been used to structure the care process for Anita (Yura & Walsh 1978). Threaded throughout is the consideration of the young person’s voice and family-centred care (NICE 2021b). Activity of living: maintaining a safe environment – pain management as per Roper, Logan, & Tierney model of care (Holland & Jenkins 2019), see Chapter 1 for further reading. An acute painful crisis is a medical emergency therefore, your initial assessment of Anita’s pain would be to assess the location and intensity of her pain using an age-appropriate pain tool and documenting her pain score on a pain assessment chart. Anita self-reported a pain score of 8 out of 10 and as pain is a subjective experience, self-report is preferred whenever possible (Twycross et al. 2015). According to the World Health Organisation (WHO) pain ladder (2018), her pain is classified as severe therefore, the primary goal for the children’s nurse is to administer strong fast acting opioids within 30 minutes (NICE 2012). Consider analgesia already taken before her presentation and document vital signs including sedation score (AVPU) on a national PEWs chart which also allows the nurse to assess the adequacy of analgesia regularly. Common triggers such as sudden temperature changes, infection, dehydration, and stress/over exertion are likely contributors to a child with SCD presenting in pain. The children’s nurse should be aware of possible triggers including other acute complications that could present during a painful crisis for example, acute chest syndrome, as this will enable prompt identification and management. Most painful crisis are managed at home; however, uncontrollable pain requires hospital management. Therefore, children’s nurses should never underestimate pain experienced. Understanding CYP pain scores, including factors that may influence pain expression for example, past experiences is key to achieving pain control (Collins et al. 2020). Pain management is an essential part of Anita’s care and should include SMART goals and monitoring tools to ensure interventions are effective. A proactive approach should be taken therefore, children’s nurses must escalate sub-therapeutic doses of analgesia prescribed, advocating for dose optimisation, to prevent further escalation of her pain. Recognition of potential side effects from opioids is key to avoiding a deterioration of Anita’s health which moves away from the desired goal. The goal is to keep Anita within the ‘analgesic corridor’ therefore, analgesia should be administered within 30 minutes of arrival to A&E with an aim to achieve pain control within 60 minutes (regular evaluation of implementation is essential. See Figure 37.1 and Box Activity 37.4. FIGURE 37.1 Analgesic corridor. Image adapted from Macintyre and Stephan (2007).
CHAPTER 37
Sickle Cell Disease
INTRODUCTION
Question 1. What Is Sickle Cell Disease?
Question 2. Genetic Inheritance, Carrier, and Screening.
Question 3. Diagnosis Treatment and Long-term Management of SCD.
ANSWER TO QUESTION 1
Pain Management
Assessment
Planning
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


