Sarcomas
Chondrosarcoma
INCIDENCE
• Primary bone cancers are extremely rare, accounting for less than 0.2% of all cancers.
• Chondrosarcoma is the second most common form of bone cancer, accounting for 30% of bone cancers in the United States.
• An estimated 4,720 new cases will be diagnosed in 2007 in the United States and 790 people will die from the disease.
• Chondrosarcoma is usually found in middle-aged and older adults.
DIAGNOSTIC WORKUP
Imaging of the primary lesions includes the following:
HISTOLOGY
G1: Well differentiated—low grade
G2: Moderately differentiated—low grade
G3: Poorly differentiated—high grade
TREATMENT
• High-dose photons are used to treat unresectable high and low-grade lesions.
• Proton beam radiation therapy





• Local recurrence or relapse should be treated with wide excision with or without radiation therapy depending on margin status.
• Radiation therapy should be considered after excision with positive surgical margins. Negative surgical margins should be observed.
• Unresectable recurrences are treated with either conventional or proton beam radiation therapy.
• Surgical excision is an option for systemic relapse of a high-grade lesion.
• Patients should also be considered for participation in a clinical trial.
PROGNOSIS
Late metastases and recurrences after 5 years are more common with chondrosarcoma than with other sarcomas.
Bovee J.V., Cleton-Jansen A.M., Taminiau A.H., et al. Emerging pathways in the development of chondrosarcoma of bone and implications for targeted treatment. Lancet Oncology. 2005;6:599–607.
Bruns J., Elbracht M., Niggemeyer O. Chondrosarcoma of bone: an oncological and functional follow-up study. Annals of Oncology. 2001;84:93–99.
Fiorenza F., Abudu A., Grimer R.J., et al. Risk factors for survival and local control in chondrosarcoma of bone. Journal of Bone and Joint Surgery British. 2002;84:93–99.
Hug E.B., Slater J.D. Proton radiation therapy for chordomas and chondrosarcomas of the skull base. Neurosurgery Clinics of North America. 2000;11:627–638.
Lee F.Y., Mank H.J., Fondren G., et al. Chondrosarcomas of bone: an assessment of outcome. Journal of Bone and Joint Surgery American. 1999;81:326–338.
Mankin H.J., Cantley K.D., Lipielo L., et al. The biology of human chondrosarcoma, I: description of cases, grading, and biochemical analyses. Journal of Bone and Joint Surgery American. 1980;62:160–176.
Mankin H.J., Cantley K.D., Schiller A.L., et al. The biology of human chondrosarcoma, II: variation in chemical composite among types and subtypes of benign and malignant cartilage tumors. Journal of Bone and Joint Surgery American. 1980;62:176–188.
Terek R.M. Recent advances in basic science of chondrosarcoma. Orthopedic Clinics of North America. 2006;37:9–14.
Ewing’s Sarcoma
DIAGNOSTIC WORKUP
If ESFT is suspected as a diagnosis, the patient should undergo a complete staging before biopsy.
• Imaging of the primary lesions
• Laboratory values most often elevated in Ewing’s sarcoma:
• Cytogenetic analysis of the biopsy specimen to evaluate the t(11;22) translocation
• Bone marrow biopsy should be considered to complete the workup.
HISTOLOGY
G1: Well differentiated—low grade
G2: Moderately differentiated—low grade
G3: Poorly differentiated—high grade
CLINICAL STAGING
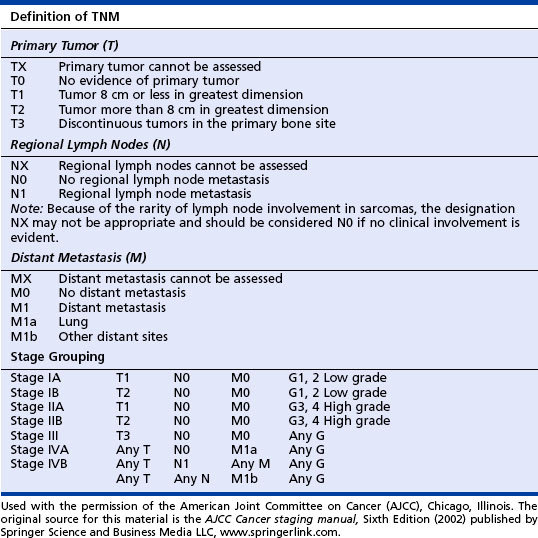
The American Joint Committee on Cancer (AJCC) TNM Staging System for Bone Sarcomas is used for all bone sarcomas. See the table on page 122 for stages of Ewing’s sarcoma according to the AJCC system.
TREATMENT




• Radiation therapy with or without surgery followed by chemotherapy or best supportive care is recommended for unresponsive or progressive disease.
• Investigational approaches should be considered in patients with recurrent and metastatic disease.
Avigad S., Cohen I.J., Zilberstein J., et al. The predictive potential of molecular detection in the nonmetastatic Ewing family of tumors. Cancer. 2004;100:1053–1058.
Bernstein M., Cover H., Paulsen M., et al. Ewing’s sarcoma family of tumors: current management. Oncologist. 2006;11:503–519.
Cotterill S.O., Ahrens S., Paulussen M., et al. Prognostic factors in Ewing’s tumor of bone: analysis of 975 patients form the European Intergroup Cooperative Ewing’s sarcoma study group. Journal of Clinical Oncology. 2000;18:3108–3114.
De Alva E., Gerald W.L. Molecular biology of the Ewing’s sarcoma/PNET. Journal of Clinical Oncology. 2000;18:204–213.
DeAlva E., Kawai A., Healy J.H., et al. EWS-FLI1 fusion transcript structure is an independent determinant of prognosis in Ewing’s sarcoma. Journal of Clinical Oncology. 1998;16:1248–1255.
Denny C.T. Gene rearrangements in Ewing’s sarcoma. Cancer Investigation. 1996;14:83–88.
Hawking D.S., Schuetze S.M., Butrynski J.E., et al. 18F-fluorodeoxyglucose positron emission tomography predicts outcome for Ewing sarcoma family of tumors. Journal of Clinical Oncology. 2005;23:8828–8834.
Zoubeck, A., Codkhonr-Dworniczak, B., Delattre, O., et al. Does expression of different EWS chimeric transcripts define clinically distinct risk groups of Ewing’s tumor patients? Journal of Clinical Oncology 12, 1245-1251.
Kaposi’s Sarcoma
ETIOLOGY AND RISK FACTORS
SIGNS AND SYMPTOMS
• Red to brown to purple growth on the skin—found mostly on the legs and face
• Mouth and throat lesions, mainly on the roof of the mouth
• The throat may be tender and sore and may bleed. Eating, breathing, or swallowing may be uncomfortable.
• Lesions can contribute to dental problems, including tooth loss.
• Possible abdominal discomfort
• Lymphedema is seen in some people; it may appear with or without skin lesions.
• May develop in the lung and produce cough, shortness of breath, and fever



