CHAPTER 18 Recognising and managing shock
Introduction
Despite advances in diagnosis, treatment and management, shock remains one of the leading causes of death in the acutely ill patient (Dellinger et al 2008). It is important that nurses from all areas of clinical practice have the knowledge required to identify patients at risk and ensure timely and appropriate intervention is carried out. Identification of the acutely deteriorating patient is a key function of the nurse. The timely and accurate recording of vital signs is an essential part of this identification. Allied to this is the appropriate escalation of management to the appropriate personnel (NICE 2007). The aim of this chapter is to help you understand the pathophysiology of shock and the associated care as this is the responsibility of all nurses.
The pathophysiology of shock
Circulatory homeostasis exists when the circulating blood volume and the vascular tone of blood vessels are in dynamic equilibrium (Marieb & Hoehn 2007, pp. 733–742). Shock is a state in which tissue perfusion is inadequate to maintain the supply of oxygen and nutrients necessary for normal cellular function and disequilibrium ensues. If there is an inadequate supply of oxygen and nutrients to two or more organs this is known as multi-organ dysfunction and indicates that the patient is at significant risk of becoming critically ill if appropriate treatment is not given immediately (Richards & Edwards 2003).
Tissue perfusion is reflected by the arterial blood pressure (Marieb & Hoehn 2007, pp. 723–733). Hypotension (systolic blood pressure below 90 mmHg or a mean arterial pressure of below 70 mmHg) is a result of either inadequate cardiac output or low systemic vascular resistance. Glomerular filtration (see Ch. 8) is dependent on an adequate blood pressure and in the presence of hypotension the urine output will drop (Marieb & Hoehn 2007, pp. 1007–1011). Inadequate perfusion to the brain may induce a change in behaviour which could be manifested through decreased arousal or agitation. Decreased arousal can be assessed using a simple mnemonic, AVPU – Alert; responds to Voice; responds to Pain; Unconscious (McNarry & Goldhill 2004). In order to maintain cardiac output, in the presence of hypotension, the heart rate will rise (Marieb & Hoehn 2007, pp. 698–705). To maintain adequate oxygenation it is likely that the respiratory rate will also increase. Thus shock is evident in the presence of:
Shock may be the result of a reduced amount of blood returning to the heart (inadequate ventricular filling), an inability of the heart to contract with sufficient strength (reduced ventricular contractility) or a decrease in the resistance to blood flow caused by dilation of the blood vessels (decreased systemic vascular resistance). Shock may be classified as follows (Bridges & Dukes 2005):
The stages of shock
Shock represents a complex set of physiological reactions which comprise activation of the sympathetic nervous and immune systems, hormonal response and metabolic derangement (Marieb & Hoehn 2007, pp. 740–742). To interpret the signs and symptoms that present and the appropriate management of shock it is necessary to understand these underlying physiological reactions. These can be divided into four stages:
The compensatory stage
In this stage physiological adaptations occur in an attempt to overcome the original problem, e.g. hypovolaemia. The compensatory stage strives to maintain cardiac output, which comprises stroke volume and heart rate (CO = SV × HR, see Ch. 2). In the absence of direct measurement of cardiac output, blood pressure and central venous pressure are used as surrogates in the assessment of cardiac output. The normal cardiac output is approximately 5 L, as is circulating volume. When circulation becomes inadequate due to a reduction in circulating volume, diminished myocardial contractility or massive vasodilatation, various mechanisms are activated in response to hypotension, hypoxaemia, acidosis or a combination of these.
Sympathetic nervous system
Hypotension leads to decreased stimulation of the aortic and carotid sinus baroreceptors (Marieb & Hoehn 2007, pp. 727–732). This reduces impulses to the vasomotor centre and thus activates vasoconstriction. Stimulation of the sympathetic nervous system occurs, resulting in activation of the stress response. Catecholamines, namely adrenaline and noradrenaline, are released from the adrenal medulla (Marieb & Hoehn 2007, pp. 631). The stimulation of alpha-adrenergic receptors by the catecholamines results in vasoconstriction of the skin, kidneys, gastrointestinal tract and other organs together with a rise in heart rate (Marieb & Hoehn 2007, pp. 543). This demonstrates an attempt to preserve the blood supply to the heart and brain. Conversely, beta-adrenergic receptors stimulate vasodilation in the lungs and skeletal muscle. Vasoconstriction may initially restore the arterial blood pressure to normal, but peripheral resistance will be raised, making the myocardium work harder to maintain cardiac output. Urine output and peristalsis will decrease, and the individual’s skin will become pale and cool. Sympathetic nervous system stimulation will also result in increased respiratory rate and depth, dilated pupils and increased sweat gland activity, causing the ‘clammy’ skin typically found in all forms of shock, other than early septic and anaphylactic shock.
Immune response
The disorder to blood flow caused by generalised vasoconstriction results in the production of proinflammatory mediators by neutrophils and macrophages (Marieb & Hoehn 2007, pp. 657–660). These include interleukins 1, 6 and 8, TNFα and prostaglandins (Greenwood & Murgo 2007). They are designed to fight foreign antigens and promote wound healing (Marieb & Hoehn 2007, pp. 790–798). At the same time anti-inflammatory mediators are produced to ameliorate the effect of these. If homeostasis is not regained then the proinflammatory mediators proliferate and damage to the endothelium occurs (Marieb & Hoehn 2007, p. 120). Neutrophils adhere to the damaged endothelium and produce more proinflammatory mediators which in turn activate the clotting cascade through the production of platelet activating factor. Small clots form which, in addition to vasoconstriction, disrupt blood flow through the organs resulting in reduced delivery of oxygen and glucose to the cells. The composition of endothelial cells is also disrupted by proinflammatory mediators and the movement of water and plasma proteins from the intravascular to the extravascular space becomes apparent in the formation of oedema.
Hormonal response
Adrenaline (epinephrine) stimulates the anterior pituitary gland to release adrenocorticotrophic hormone (ACTH), which causes the adrenal cortex to release glucocorticoids such as hydrocortisone and mineralocorticoids such as aldosterone (Marieb & Hoehn 2007, pp. 616, 626–629).
Aldosterone
increases reabsorption of sodium and chloride by the kidney. To maintain electrolyte balance at the renal tubular level, the excretion of potassium and hydrogen ions is increased. Thus hypokalaemia can occur. A higher concentration of sodium chloride raises the serum osmolality stimulating the hypothalamic osmoreceptors. In turn this leads to the release of antidiuretic hormone (ADH) from the posterior pituitary gland (Marieb & Hoehn 2007, pp. 627–628).
ADH
stimulates an increase in renal tubular water reabsorption, in an attempt to augment circulating volume and blood pressure whilst restoring normal serum osmolality (Marieb & Hoehn 2007, pp. 617–619). Urine output is diminished.
Renin
is secreted by the kidney in response to secretion of noradrenaline by the adrenal medulla, which results in renal artery vasoconstriction. In the circulation, renin reacts with angiotensinogen, producing angiotensin I. This is converted by an enzyme in the lungs to angiotensin II, which causes venous constriction and increases aldosterone release, thus leading to increased fluid and sodium retention, increased blood volume and therefore increased venous return, blood pressure and renal perfusion (Marieb & Hoehn 2007, p. 628).
The progressive stage
If tissue perfusion is not restored and blood flow through the organs remains erratic then the supply of oxygen and glucose to the cells will be inadequate to produce sufficient adenosine triphosphate (ATP) (Marieb & Hoehn 2007, p. 57). The sodium pump will fail and the amount of intracellular calcium increase. Anaerobic metabolism by the cell, which results from an inadequate oxygen supply, will increase the production of lactic acid resulting in a metabolic acidosis (pH <7.35) (Edwards 2008).
The refractory stage
In this stage pathophysiological processes are set in motion that cannot be arrested or reversed. Death is imminent. Continuing circulatory collapse, an inability to restore circulating blood volume, increasing metabolic acidosis and the formation of micro-emboli will all contribute to decreased tissue perfusion (Marieb & Hoehn 2007). Inadequate ventilation will lead to increasingly inadequate oxygenation. Renal failure will contribute to increasing metabolic abnormalities, and vital centres in the brain will eventually cease to function due to ischaemia and hypoxia. If the refractory stage is reached, shock is irreversible and death will occur.
Types of shock
The following sections describe the distinguishing features of hypovolaemic shock, cardiogenic shock, obstructive shock and the different forms of distributive shock (Skinner & Jonas 2007). It is essential that deterioration in the patient’s condition is communicated effectively and escalated to experienced staff. This is addressed in a later section of this chapter. (See Boxes 18.1, 18.2.)
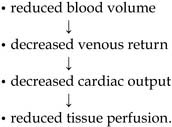
Hypovolaemic shock
The physiological implications of hypovolaemia are:
Recognising hypovolaemic shock
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


