Chapter 32 Pharmacology and prescribing
Learning outcomes for this chapter are:
1. To discuss the scope of midwifery practice in relation to prescribing medicines and the legislation associated with the prescription of medicines
2. To revise pharmacological concepts that inform the prescription of medicines and discuss the safety of drugs in pregnancy
3. To revise pharmaceutical medicines commonly used in maternity care
4. To review the use of antimicrobials in relation to maternity care and midwives’ responsibility for minimising bacterial resistance
5. To examine the evidence in relation to accurate assessment, diagnosis and prescription of medicines related to the midwifery scope of prescribing
6. To be able to accurately prescribe appropriate medicines as part of autonomous midwifery practice.
minimum effective concentration
non-steroidal anti-inflammatory drugs (NSAIDs)
pharmacodynamics, pharmacokinetics
Limitations of this chapter: The authors assume that readers have prior pharmacology knowledge and a comprehensive knowledge of normal and complicated childbirth. The discussion in this chapter is limited to pharmaceutical medicines and does not cover the use of complementary or non-pharmaceutical therapies. The authors acknowledge that prescription of medicines is always evolving according to research, availability of pharmaceuticals and cost. Midwife prescribers have an obligation to maintain a current knowledge of the safety of medicines in pregnancy and lactation, and of preferred pharmaceuticals for particular conditions.
SCOPE OF MIDWIFERY PRESCRIBING
Dependent prescribing is prescribing according to standing orders or protocols where authority remains with the medical practitioner, whereas independent prescribing occurs when the healthcare practitioner has the legal authority to issue prescription medicines. The report of the Maternity Services Review in Australia (Commonwealth of Australia 2009) indicated support for an expanded role for the scope of midwifery practice. Legislative changes in Australia through the Health Legislation Amendment (Midwives and Nurse practitioners) Act 2010 will enable midwives to claim from the Medicare Benefits Schedule and the Pharmaceutical Benefits Scheme. Further detail in relation to this extended role for Australian midwives is covered in Chapters 1 and 13 of this textbook but full details of implementation are not yet clear. Midwives in New Zealand undertake dependent and independent prescribing, and the Midwifery Council of New Zealand (MCNZ) expects all registered midwives to be able to demonstrate competence in independent prescribing. Competency 2.13 (MCNZ 2007) indicates that a midwife must demonstrate the ability to prescribe, supply and administer medicine, vaccines and immunoglobulins safely and appropriately within the midwife’s scope of practice and the relevant legislation.
Midwives in New Zealand gained lawful prescribing rights through the passage of the Nurses Amendment Act 1990, which included amendments to the Misuse of Drugs Act 1975 and the Medicines Act 1981. There is no defined list of medicines a midwife may prescribe, but the limits as to when a midwife can prescribe are set out in an amendment to Regulation 39 of the Medicines Regulations 1984, which states: ‘No registered midwife shall prescribe any prescription medicine otherwise than for antenatal, intrapartum and postnatal care’ and the quantity of any prescription medicine shall not exceed a three-month supply. The Misuse of Drugs Act 1975 permits midwives to prescribe pethidine, the only controlled drug that midwives can prescribe.
Following the law changes above, the New Zealand Department of Health (1990) circulated a guide to the Nurses Amendment Act 1990 stating that midwifery prescribing would not include the treatment of underlying medical conditions such as asthma or hypertension, and that ‘it would also not include the prescribing of medicines such as antibiotics or oral contraceptives’ (Department of Health 1990, p 5). However, caseloading midwives found the latter restriction untenable. It imposed additional expense on women to consult a doctor in order to obtain medicines such as the progestogen-only contraceptive pill, or antibiotics for an uncomplicated urinary tract infection. The New Zealand College of Midwives (NZCOM) challenged the restrictions on prescribing, and during 1995 the Ministry of Health agreed that there was no legal basis for restricting the prescription of antibiotics and oral contraceptives and that it may indeed be appropriate for these to be prescribed by midwives during the course of providing antenatal, intrapartum and postnatal care (K Guilliland, personal communication 1998).
The NZCOM Consensus Statement on Prescribing (NZCOM 2002) notes that midwives can prescribe for conditions commonly associated with uncomplicated pregnancy, labour and the postnatal period, up to six weeks after the birth of the baby (Campbell 2003). Midwives need to have knowledge of the effects, side-effects and contraindications of the drugs prescribed and know to prescribe within their knowledge and expertise. The NZCOM strongly discourages the use of analgesics or sedatives during labour at home. The necessity for these types of medications is an indication for transfer to hospital. If narcotics are prescribed for use in labour, these must be discussed with the woman and the midwife must have the equipment and skills necessary to cope with the effects of this medication.
SAFETY OF DRUGS DURING PREGNANCY AND LACTATION
Thalidomide is well known as the prototype for teratogens (de Santis et al 2004; Kyle 2006). Thalidomide prescribed to pregnant women in the 1960s to relieve nausea and vomiting resulted in babies born with limb malformations, and this catastrophic effect shook the perception that the placenta provides a barrier to drugs (Gardiner 2002). Drug licensing was less rigorous in the 1960s, and it was the compilation of case reports that established the link between thalidomide and infant deformities (Freyer 2008). Drug use during pregnancy should be restricted according to necessity, and to the use of drugs for which prior clinical evidence is available. The first trimester of pregnancy is generally considered the most critical period for teratogenic effects. After the first 11 weeks, exposure to teratogens will generally have an effect on the nervous system, gonadal tissue (due to slower development than other organs), or general growth restriction (Hansen et al 2002). Vigilance is also necessary when midwives prescribe medicines during lactation. Resources should be utilised to ascertain the safety of medicines.
Studies show that many women use drugs during pregnancy. Schirm and colleagues (2004) evaluated drug types by prescription for 7500 Danish women. The authors reported that 69% of the women used a drug during pregnancy that was not folic acid, an iron preparation or vitamins. Drugs commonly used included dermatological and gynaecological preparations, lactulose, salbutamol and paracetamol. Andrade and colleagues (2004) similarly reported that 64% of women in a study in the United States (total study n = 98,182) used a drug other than a vitamin or mineral supplement during pregnancy, with the most frequently used drugs being oral anti-infective medicines.
There appears to be a prevalence of drug use during pregnancy, and therefore it is important that midwives caution women about safety of medications during pregnancy, particularly with the availability of over-the-counter pharmaceuticals. The Swedish, Australian and US Federal Drug Administration drug classification systems are well known for classifying drug safety, using the codes A, B, C, D and X. However, evaluation showed that only 26% of drugs common to all three systems were placed in the same risk category (Freyer 2008; Schirm et al 2004). These differences in categories cause confusion for the prescriber. The Australian categorisation of drugs and safety during pregnancy provides additional narrative information to guide the prescriber. Drugs are categorised to assist the prescriber to select the ‘safest’ known medicine appropriate for a particular condition.
Reflective exercise
Refer to the following information and then answer the questions below.
Prescribing medicines in pregnancy: an Australian categorisation of risk of drug use in pregnancy (Department of Health and Ageing, Therapeutic Goods Administration 1999 with updates to 2007. Online: www.tga.gov.au/docs/html/mip/medicine.htm).
Questions
1. Explain in your own words the drug categories A–X and the relevance of these categories to pregnancy.
2. List the drugs commonly used during pregnancy and childbirth, and the category of each drug.
3. Review your personal habits regarding the use of pharmaceuticals in relation to pain, flu or mild infections.
4. Discuss your personal biases with regard to drug use during pregnancy and childbirth (e.g. women experiencing vomiting, women wishing to use antacids or laxatives regularly).
PRINCIPLES OF PHARMACOLOGY
Pharmacokinetics
Absorption
In most cases, when a drug is administered to a person it has to be absorbed into the bloodstream before the molecules can be distributed around the body to the site of action. Absorption is a complex process, as age, body mass, activity and fullness of the stomach can affect both the amount and the rate at which drugs are absorbed. Decreased gastrointestinal motility can increase or decrease drug absorption during pregnancy, and vomiting could mean that the drug is not absorbed at all (Bryant et al 2007; Shargel 2004). Once the drug is dissolved in the gastrointestinal tract, the molecules can be absorbed. Because plasma membranes primarily consist of lipid substances, drug molecules generally need to be lipophilic (having an affinity with lipids) to pass through into the capillaries (Bullock et al 2007; Shargel 2004).
Distribution
Hepatic portal system
As with most other digested substances, drugs are absorbed from the intestine into the hepatic circulation via the portal vein. Therefore, before the drug molecules can reach the general circulation they must pass through the liver. This is known as the hepatic first pass. At this point, a proportion of drug molecules can be metabolised and excreted by the liver. Drugs that have a significant proportion of molecules metabolised in this way are said to have a high hepatic first pass (Bullock et al 2007; Holland & Adams 2007).
Some of the metabolites from the liver are excreted into the small intestine via the bile duct. Bile salts are excreted in this way. The body uses intestinal bacteria to recycle bile salts. The bacteria de-conjugate the bile salts, allowing them to be reabsorbed from the small intestine so that they can be conjugated again in the liver, excreted back into the small intestine and used once again in the normal digestion process. This is known as the enterohepatic cycle. A proportion of drug molecules metabolised in the liver on the hepatic first pass are excreted into the intestine, where they are also recycled by undergoing the process of de-conjugation, reabsorption from the gut and then passing through the hepatic circulation a second time. Oestrogen in the combined oral contraceptive pill is an example of a drug that is affected in this way (Bryant et al 2007; Bullock et al 2007; Holland & Adams 2007).
General circulation
Once the molecules reach the general circulation, they are distributed to the site of action. Highly soluble drugs are carried in the plasma as free molecules in solution. Less-soluble drugs are carried partially bound to plasma protein molecules. Only drug molecules that are free in solution are pharmacologically active. They cannot bind to the receptor sites in the body while still bound to the serum protein. The levels of free and protein-bound drug molecules are in equilibrium. This means that drug molecules that are bound to protein molecules are released and become active as the free molecules are used. Therefore drugs that are distributed in the body partially bound to protein molecules take longer to be released to the receptor sites. The partially bound drug is pharmacologically active for longer than a highly soluble drug that has all its molecules available to the receptor sites and all its molecules available for metabolism by the liver at the same time.
The increase in maternal plasma volume up to 50% within the first 10 weeks of gestation (Weiner et al 2005) and increase in body fat during pregnancy can affect drug distribution. This does not affect drug dosing except for loading doses, where a higher dose might be required (Gardiner 2002). The increase in body fat might affect drugs that are deposited in fatty tissue, resulting in a decrease in plasma concentration.
Box 32.1 Barriers to distribution
Blood–brain barrier
The brain is protected from harmful substances because it has a different capillary structure from that of the rest of the body. The junctions between endothelial cells are narrower, reducing the number of pores available. Further, a web of tightly connected glial cells forms a fatty layer between the capillaries and the brain tissue. Only material that is very fat soluble or is actively transported can cross the blood–brain barrier (Bryant et al 2007; Bullock et al 2007; Shargel 2004).
Placenta
The poor protection afforded by the placenta is the primary reason that drugs are generally contraindicated in uncomplicated pregnancies. When a pregnancy is complicated by a concurrent medical condition, medications are prescribed with caution. In these cases, the obstetrician assesses the risk/benefit of medication in each situation. Some anticonvulsants commonly used to control epilepsy in non-pregnant women pose a significant risk to a growing baby. The woman’s medication may be changed to a drug that poses less risk to the baby for the duration of the pregnancy. In other medical conditions, the dose(s) of drug used to control the medical condition may need to be adjusted and plasma concentrations monitored (Bryant et al 2007).
concentrations such as phenytoin (Begg 2008; Bryant et al 2007; Shargel 2004). Phenytoin concentrations during pregnancy are extremely complicated and specialist interpretation of serum levels is necessary (Freyer 2008).
Metabolism and excretion
If the drug molecules remain lipophilic they will be reabsorbed at the nephron tubules or returned to the gut from the liver. Therefore drugs are metabolised in the liver to make them water soluble (polar) so they can be excreted from the body. Two types of enzyme are involved in metabolism of drugs. The first type (sometimes called phase I) modifies the drug by chemical processes such as oxidation, reduction or hydrolysis to make water-soluble metabolites. The cytochrome P450 family of enzymes found in abundance in the hepatocytes is responsible for most of these reactions. Induction or inhibition of drug metabolism during pregnancy is dependent on the specific P450 enzymes involved. During pregnancy, metabolism of caffeine is decreased, whereas that of phenytoin is increased (Begg 2008; Bryant et al 2007).
The second type of enzyme (phase II) conjugates the drug molecules, or a phase I metabolite, with a polar molecule such as glucuronic acid, rendering the molecule water soluble for excretion. Conjugation enzymes belong to the transferase family. Midwives will be familiar with the physiological process in neonates whereby excess lipophilic bilirubin molecules are conjugated to water-soluble molecules by the enzyme glucuronyl transferase in the liver (Begg 2008; Bryant et al 2007). Drugs are mainly excreted in the bile and urine; other routes include the lungs, sweat and saliva, and breast milk in lactating women (Bullock et al 2007). Drugs that are very water soluble are excreted virtually unchanged into the urine. This can be helpful when treating bacterial infections of the lower urinary tract (Bryant et al 2007).
Drug clearance may be increased during pregnancy due to an increase in renal and liver blood flow (Freyer 2008). The glomerular filtration rate increases from early pregnancy and remains elevated throughout the pregnancy; therefore increased doses are required for drugs such as beta-lactam antibiotics, which are eliminated through the renal system (Begg 2008; Gardiner 2002).
Bioavailability
Bioavailability is the amount of drug that is available to the receptor sites. When injected intravenously, 100% of the drug is available; but when given orally some of the drug is lost during the absorption and hepatic first pass processes. Therefore, a smaller dose is usually required when the intravenous route is used (e.g. doses of narcotics). Antibiotic therapy is the most common exception, as these drugs act directly on bacteria, not on body cells (Bryant et al 2007; Bullock et al 2007).
Plasma concentration
The time taken after a dose of a drug to reach maximum plasma concentration is affected by absorption, distribution, metabolism and elimination rates. Drugs given orally take longer than those administered intravenously to reach peak plasma concentration. The plasma concentration is related to the therapeutic effect (Begg 2008).
Therapeutic range
In order to produce a clinical effect, a certain level of drug needs to be present in the general circulation. This level is called the minimum effective concentration (MEC) and is different for each drug. For all drugs there is also a level beyond which the drug will produce adverse effects on the person; this is known as the maximum safe concentration (MSC). The range between the MEC and the MSC that is associated with drug efficacy is known as the therapeutic range. Most drugs have a wide therapeutic range, which means that the serum levels will not normally rise to toxic levels when the usual size and frequency of dose is taken. However, a significant number of drugs, especially anticonvulsants and antibiotics from the aminoglycoside family (e.g. gentamycin) have a narrow therapeutic range. This means that it is possible for serum levels to rise to toxic levels even with normal dose and frequency regimens. People being treated with drugs that have a narrow therapeutic range need to have the serum levels of the drug regularly monitored so that, if necessary, the dose can be adjusted to maintain effectiveness without causing toxicity (Begg 2008; Bryant et al 2007; Bullock et al 2007).
Half-life
The length of time for which a drug is clinically effective in the body is called its half-life. Half-life is the length of time it takes to reduce the amount of drug in the circulation by half. For example, 500 mg of a drug that has a half-life of four hours will reduce to 250 mg four hours after administration, to 125 mg after another four hours, and so on. Although there are still active molecules of the drug in the body after four hours (in this example), the clinical effectiveness of the drug has been significantly reduced. This principle guides recommendations for the frequency of doses. Doses are given at the half-life interval (e.g. four-hourly in the above example) (Bryant et al 2007; Bullock et al 2007; Shargel 2004).
Steady-state concentration
The aim of dosing regimens is to achieve a steady-state concentration—that is, to maintain a constant concentration of the drug in the plasma that is consistent with a therapeutic response in the person (Fig 32.1). When a steady state is reached, the maintenance dose rate is equal to the elimination rate (i.e. it is in equilibrium) (Bryant et al 2007; Shargel 2004). Drugs are given at half-life intervals to reach a steady-state concentration: ‘In general it takes 3 to 5 half lives to reach the desired steady state’ (Bryant et al 2007, p 122). To avoid the problem of delay when the half-life is long or rapid and treatment is imperative, a loading dose is often given.
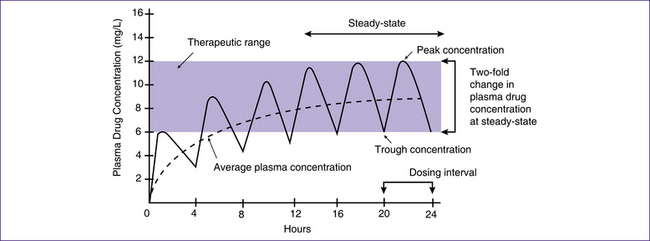
Figure 32.1 Achieving a steady state. The drug in this example has a half-life of four hours
(based on Bryant et al 2007)
While loading doses are often twice the ongoing dose, the desired plasma concentration and the apparent volume of distribution for individual drugs are taken into account (Shargel 2004). The initial dose is sometimes given parenterally, followed by oral administration of maintenance doses.
Pharmacodynamics
Pharmacodynamics is the term used to describe the action of a drug on the body. Nearly all drugs act on receptors in the body (antimicrobials act on microorganisms) by binding to a protein. Most drugs have some selectivity—that is, they ‘see’ or ‘fit’ particular receptors as targets. Generally, drugs act on four main types of regulatory proteins: carriers, enzymes, ion channels and receptors (Bryant et al 2007; Holland & Adams 2007).
Carriers transport ions and small, poorly fat-soluble molecules across plasma membranes. Examples are carriers that uptake noradrenaline and serotonin at the nerve terminals. Drugs that target carriers usually inhibit carrier-mediated uptake of such transmitters (e.g. tricyclic antidepressants) (Bryant et al 2007).
Enzymes are biological catalysts that control cellular biochemical reactions. Drugs that interact with enzymes inhibit or alter the biochemical reaction. The sulfonamide group of drugs, which includes the drug trimethoprim, are an example (Bullock et al 2007).
Ions are transported in and out of cells through specific channels in the plasma membrane so that the electrochemical gradient across the plasma membrane can be maintained. Drugs that interact with receptors to block the ion channels are widely used. These drugs generally have a high selectivity. Nifedipine, used for the control of hypertension, for example, is a calcium-ion-channel blocker that affects the arterioles but has little effect on cardiac muscle and no effect on the transport of other ions such as sodium (Bryant et al 2007; Bullock et al 2007).
Receptors are specific proteins that span plasma membrane. They are engaged in ‘chemical signalling between and within cells’ (Bryant et al 2007, p 95). Drug molecules bind with receptors in a ‘lock and key’ effect. Drugs that temporarily bind to receptors and stimulate the cell to carry out its normal biochemical actions are known as agonists, while those that bind to the receptors to temporarily block or inhibit the normal cell process are referred to as antagonists. Drugs called ‘blockers’ are antagonists. The beta-blocker labetalol is an example (Bullock et al 2007; Holland & Adams 2007).
ANTIMICROBIALS
Box 32.2 Prescription guidelines
To ensure accurate prescription of a pharmaceutical medicine:
• Use accurate physical examination and diagnostic skills.
• Consider whether a pharmaceutical is appropriate for the condition.
• Have comprehensive knowledge of ‘commonly’ used drugs, including action/effect, and communicate this information to the woman.
• Ensure that the treatment is regarded as safe during pregnancy or during lactation.
• Consider the evidence in relation to the clinical scenario.
• Prescribe the appropriate drug, dose, frequency and duration of treatment.
• Monitor for any adverse reactions and refer accordingly.
• Do follow-up culture and sensitivity and alter drug if necessary.
• Be able to research information about drugs used less commonly.
• Have a low threshold for consultation with an obstetrician or general medical practitioner.
(Source: adapted from Banning 2004 and Lethard 2001)
reinforce information about judicious use of antibiotics, recognising adverse effects and preventing the development of resistant bacteria arise in the course of midwifery practice.
Midwives do not independently prescribe for infections that require intravenous antibiotics; referral should be made to an obstetrician. Midwives would not prescribe for medical conditions such as pyelonephritis or prophylactic antibiotics for cardiac conditions. Neonates with symptoms suggestive of systemic infection should be promptly referred to the paediatric service (or a medical practitioner in areas without a paediatric service). Neonates have immature liver enzymes and renal system, and therefore have limited ability to metabolise and excrete drugs. They can also become seriously ill very quickly. Referral should not be delayed. There is emerging evidence of a link between early exposure to antibiotics and the development of recurrent wheeze in the first two years of life (Kummeling et al 2007).
Antimicrobials are groups of drugs that destroy or restrict the proliferation of infectious microorganisms. They work by inhibiting cell wall synthesis, disrupting plasma membrane permeability, interfering with the organism’s metabolic processes, or by inhibiting protein synthesis in the organism (Bryant et al 2007; Bullock et al 2007).
Bacteriocidal antimicrobials destroy the organism and bacteriostatic drugs hinder bacterial growth, giving the body a longer period in which to mobilise its own defences. Antibiotics are used to treat bacterial infections. Other antimicrobials include antiviral, antifungal and antimycobacterial agents (Holland & Adams 2007).
Spectrum of activity
The number of types of organisms that are sensitive to a particular antibiotic is what determines its spectrum of activity. Antibiotics to which only a few groups of organisms are sensitive are said to have a narrow spectrum of activity, while those that are effective against a wide range of organisms are said to have a broad spectrum of activity. A well-chosen narrow-spectrum antibiotic can be very effective. An example of this is flucloxacillin, a narrow-spectrum, beta-lactamase-resistant antibiotic, effective against Staphylococcus aureus (Bullock et al 2007).
Adverse reactions
It is important to take a careful history of hypersensitivity, adverse reaction or an anaphylactic response in relation to exposure to antibiotics. There is a world of difference between adverse reactions such as diarrhoea, nausea and vomiting, candidiasis, sore mouth or tongue (New Ethicals Compendium 2004); fever and rash due to allergy; and an anaphylactic reaction. Clients do not always distinguish between types of adverse reaction. Women who have a history of hypersensitivity to any antibiotic should have their clinical notes prominently marked so that it is not overlooked. In cases of hypersensitivity, drugs should be discontinued and consultation sought. ‘Hypersensitivity reactions occur in 4–8% of patients. Anaphylaxis occurs in approximately 0.2% [of clients]’ (Lang 2004, p 25) (see Box 32.3). Midwives need to be particularly cautious regarding women with a history of penicillin allergy. A significant number of these clients will have adverse reactions or allergies to other groups of antibiotics, and hence referral to a doctor is indicated. The tetracyclines group of antibiotics cannot be used as an alternative, as they are contraindicated in pregnancy—there is risk to the fetus of teeth staining and inhibited bone growth (Bullock et al 2007).
Box 32.3 Anaphylactic shock
Signs and symptoms
• Rapid pulse, sweating, dizziness, fainting, unconsciousness, wheezing, chest tightness, difficulty breathing, coughing, itchy hives which may blend together to form larger areas of skin swelling; swelling of the lips, tongue or eyes
• Nausea, vomiting, abdominal cramps, diarrhoea
• Paleness, bluish skin colour
• Throat swelling, with a feeling of throat tightness, a lump in the throat, hoarseness or obstructed air flow.
Treatment
• Call for emergency assistance immediately and stop any intravenous antibiotic
• If unconscious, commence resuscitation
• Administer oxygen via appropriate route (8 L O2 via facemask if maintaining respiration)
• Adrenaline is the treatment of choice. The dose of adrenaline is 0.3–0.5 mg 1:1000 IM for an adult, at the first sign of a serious reaction. Adrenaline should be prescribed by a medical practitioner, paramedic or as a standing order in emergency situations. Adrenaline given subcutaneously or intramuscularly will work for about 150 minutes.
• Following the administration of adrenaline in the community, admit to hospital for observation for up to 24 hours, as occasionally the symptoms can return.
Types of antimicrobials
Beta-lactam antibiotics
These drugs are bacteriocidal substances that contain a beta-lactam ring in their molecular structure. They include the penicillins, cephalosporins, monobactams, carbapenems and beta-lactamase inhibitors (Bullock et al 2007). Some bacteria have become resistant to antibiotics by developing the capacity to produce the enzyme beta-lactamase which breaks the beta-lactam ring in the antibiotic molecule, rendering it ineffective (Holland & Adams 2007).
Penicillins
The penicillins are a large group of both natural and synthetic antibiotics. They are grouped into several categories. Benzylpenicillin or penicillin G (parenteral) and phenoxymethylpenicillin or penicillin V (oral) are clinically comparable. They have been largely replaced by broad-spectrum penicillins such as amoxycillin, which does not need an empty stomach and is more reliably absorbed (Lang 2004). Penicillin is used selectively, for example as prophylaxis for women with a history of cardiac conditions such as bacterial endocarditis or for Group B Streptococcus prophylaxis intrapartum.
Narrow-spectrum beta-lactamase-resistant penicillins
These penicillins are beta-lactamase resistant (Bryant et al 2007; Holland & Adams 2007). Antibiotics effective against Staphylococcus aureus, such as flucloxacillin, are used for lactating women when pharmacological treatment of mastitis is required (Spencer 2008). Flucloxacillin needs to be taken on an empty stomach (one hour prior to food or two hours after food), which can be problematic. An increasing number of S. aureus isolates are resistant to flucloxacillin (Wynne et al 2007).
Extended-spectrum penicillins
The spectrum of activity for amoxycillin/clavulanic acid, piperacillin/tazobactam and ticarillin/clavulanic acid is extended with the addition of the beta-lactamase inhibitors clavulanic acid or tazobactam. This makes these antibiotics effective against otherwise-resistant strains of S. aureus and some anaerobes. Of all the penicillins, piperacillin and ticarillin have the widest range of action against Gram-negative organisms. Ticarillin is no longer available in New Zealand (Bryant et al 2007; Lang 2004).
Beta-lactamase inhibitors do not have much antibacterial activity in themselves, and reaction to clavulanic acid is extremely rare. They protect the antibiotic by binding with the beta-lactamase produced by the microorganisms (Wynne et al 2007).
Cephalosporins
Cephalosporins are a family of broad-spectrum antibiotics that are classified by their generation. The generation refers to the timeframe over which each group was developed, rather than increasing improvement. Each generation has a specific effect on different groups of bacteria. First-generation cephalosporins are active against Gram-positive bacteria; second-generation drugs also target Gram-negative organisms, as do third-generation drugs, which have a longer duration and also target beta-lactamase-producing organisms. Fourth-generation cephalosporins have similarities with the third-generation drugs (Holland & Adams 2007). Most cephalosporins are only available for parenteral use. Therefore community prescribing is limited to the few oral preparations. Cephalosporins are judiciously prescribed, as they are valuable agents in the treatment of Gram-negative organisms and are more expensive than the penicillins.
In pregnancy, cephalosporins tend to have ‘shorter half lives, lower serum levels, increased volumes of distribution and increased clearance’ (Wynne et al 2007, p 646).
Oral cephalosporins are alternatives to penicillins for treatment of skin and soft tissue infections of S. aureus and streptococcal (not enterococcal) origin (Lang 2004). Cephalosporins should not be used if a person reports a serious reaction to penicillin (Bryant et al 2007; Wynne et al 2007). Adverse effects are similar to those for penicillins but hypersensitivities are much less common. Renal toxicity has been associated with earlier-generation cephalosporins (Holland & Adams 2007).
Macrolide antibiotics
The macrolides are a group of primarily bacteriostatic antibiotics with some bacteriocidal effect, that include erythromycin and azithromycin. They have a broad spectrum of activity against diverse organisms. Erythromycin is the most commonly used alternative to penicillin where there is an allergy, and is an alternative to flucloxacillin or to cephalosporins for susceptible strains of staphylococci and other Gram-positive bacteria (Holland & Adams 2007; Lang 2004). Erythromycin estolate is contraindicated in pregnancy because of drug-related hepatotoxicity. Erythromycin ethylsuccinate is safe for use during pregnancy.
Azithromycin is effective in the treatment of both Gram-positive and Gram-negative organisms. It is effective against infections by intracellular organisms such as Chlamydia (Wynne et al 2007).
Nitrofurantoin
Nitrofurantoin is a broad-spectrum bacteriocide that inactivates or alters bacterial ribosomal proteins and other bacterial cell molecules (Medsafe 2004). It is specific for the treatment of acute urinary tract infections, as it is highly soluble and approximately 65% of the drug is excreted in the urine unchanged. Nitrofurantoin is contraindicated when labour is imminent because of the possibility of haemolytic anaemia in the neonate due to immature erythrocyte enzyme systems (glutathione instability). It is also contraindicated in women with glucose-6-phosphate dehydrogenase (G6PD) deficiency due to potential haemolytic anaemia. Similarly, nitrofurantoin should not be prescribed to lactating women because of the risk to the baby (Bryant et al 2007; Medsafe 2004).
Women should not take urine alkalinisers when taking nitrofurantoin as this drug has more efficacy in an acidic environment. Antacid preparations containing magnesium trisilicate should be avoided when taking nitrofurantoin, to avoid the possibility of impaired absorption. Absorption is increased when taken with food (Medsafe 2004). Nitrofurantoin is frequently prescribed for pregnant women who have a history of, or suffer from, recurrent urinary tract infection. There is a view held by some clinicians that nitrofurantoin should be reserved for prophylaxis or therapeutics of this nature.



