Chapter 32. Non-malignant haematological disorders of childhood
Kate Khair
LEARNING OUTCOMES
• Demonstrate an understanding of the anatomy and physiology of the haematological system.
• Gain an overview of the common haematological disorders of childhood.
• Understand the role of nurses for caring for children with non-malignant haematological disorders.
• Use relevant contemporary literature to inform the nursing care of children with non-malignant haematological disorders.
Introduction
There are two aspects of haematology which form part of the normal haematological system. The first is haematopoiesis (from the Greek, haem = blood and poiesis = to make) which involves the general aspects of blood cell formation in the bone marrow, the second is haemostasis (also from Greek haem = blood and stasis = stagnation) which is the process of blood clotting. Abnormalities within either of these two systems lead to disorders which cause significant clinical symptoms of bruising, bleeding, anaemia or infection. Some of these conditions can be acute, e.g. ITP, although the majority, e.g. sickle cell disease or haemophilia, are chronic conditions, which used to be fatal in childhood but with contemporary management can also now be seen as chronic diseases of adolescence and adulthood. Children with these conditions receive nursing care at home, given by their parents (Vidler, 1999 and Burnes et al., 2008) and themselves, as well as by community nurses, in local general hospitals, and also in tertiary care centres which may be either paediatric or haematological, or both. There are regional haemoglobinopathy centres and haemophilia comprehensive care centres which provide 24-hour access and advice about treatment of children with these complex conditions.
Anatomy and physiology of the haematological system
Haemopoiesis
Haemopoiesis occurs from the first few weeks of embryo development predominantly in the liver and spleen. In later fetal life (at about 6–7 months of gestation) the bone marrow takes over and during childhood and adulthood becomes the source of blood cell production (Hoffbrand et al 2001). In children the bone marrow in all bones is active in cell production; in adolescence as bone growth ceases cell production occurs only in the sternum, vertebrae, pelvis and ribs. The sternum and pelvis are the sites most commonly used for bone marrow aspiration to establish diagnosis of bone marrow disease/dysfunction.
Haemopoiesis begins with a stem cell which gives rise to separate lineages and develops into a variety of cells which in turn become erythrocytes (red blood cells (RBC)) leucocytes (white blood cells (WBC)) and thrombocytes (platelets) (Fig. 32.1). The normal ranges for these cells are dependant upon age and sex (Table 32.1). The specific actions of these cells are discussed below.
 |
| Fig. 32.1 Haemopoiesis |
| Age | Hb (g/dL) | MCV (fL) | Neutrophils (10 9/L) | Lymphocytes (10 9/L) | Platelets (10 9/L) |
|---|---|---|---|---|---|
| Birth | 14.9–23.7 | 100–125 | 2.7–14.4 | 2–7.3 | 150–450 |
| 2 weeks | 13.4–19.8 | 88–110 | 1.5–5.4 | 2.8–9.1 | 170–500 |
| 2 months | 9.4–13.0 | 84–98 | 0.7–4.8 | 3.3–10.3 | 210–650 |
| 6 months | 10.0–13.0 | 73–84 | 1.0–6.0 | 3.1–11.5 | 210–560 |
| 1 year | 10.1–13.0 | 70–82 | 1.0–8.0 | 3.4–10.5 | 200–550 |
| 2–6 years | 11.5–13.8 | 72–87 | 1.5–8.5 | 1.8–8.4 | 210–490 |
| 6–12 years | 11.1–14.7 | 76–90 | 1.5–8.0 | 1.5–5.0 | 170–450 |
| Adult ♀ | 12.2–15.1 | 77–94 | 1.5–6.0 | 1.5–4.5 | 180–430 |
| Adult ♂ | 12.1–16.6 | 77–92 | 1.5–6.0 | 1.5–4.5 | 180–430 |
Erythrocytes
Erythrocytes are bi-concave discs which have a lifespan of approximately 120 days. They transport haemoglobin which carries oxygen around the body. As the RBC grows old it becomes fragile, the cell ruptures and the haemoglobin is broken down into haemosiderin and bile pigments which are excreted by the liver.
Leucocytes
There are five types of white cells (leucocytes): lymphocytes, monocytes, neutrophils, eosinophils and basophils. Each of the subgroups of WBC plays a different role in immune processes and are involved in inflammation, phagocytosis, allergy and healing. All of these cells have a nucleus but have a much shorter lifespan than the RBC, being measurable in the blood for a matter of hours; however WBC leave the blood and move into tissues where they act as a reservoir to fight infection where they die after a matter of a few days (Campbell et al 1995 p 610)
Thrombocytes
Thrombocytes (platelets) are the smallest cells, originating from megakaryocytes in the bone marrow, and have a lifespan of 8–10 days. They are round discs, which are able to change shape due to their structure, they adhere to each other as well as coagulation factors to cause a platelet plug at the site of damage to the blood vessel. The surface of the platelet is coated with glycoproteins which are particularly important for adhesion and aggregation and which are fundamental for the formation of the platelet plug. Platelets are instrumental in protection from and treatment of bleeding disorders
Coagulation
Coagulation is a complex cascade of actions, involving a variety of mechanisms from blood vessel wall constriction and platelet adhesion to activation of coagulation factors which adhere to the platelets to form a clot to stop bleeding. If this mechanism was left ‘unchecked’ thrombosis would occur; to prevent this, coagulation factor inhibitors ‘switch off’ the coagulation cascade once bleeding has ceased. The coagulation cascade is defined as three interdependent parts: the intrinsic, the extrinsic and the common pathways. The intrinsic pathway begins with ‘contact activation’ with factor XII becoming activated, this converts factors XI, IX, and VIII into their active parts which work with factor X to initiate clot formation. At the same time the extrinsic pathway generates a ‘thrombin burst’ when tissue factor is released at the site of injury: activated factor VII circulates in high levels, in turn further activating factors VIII, and factors V and X, within the common pathway where finally thrombin acts to convert fibrinogen to fibrin and a haemostatic plug is formed.
With the exception of von Willebrand factor (which is produced in the endothelium) coagulation factors are produced or synthesised by the liver, therefore liver disease can present with and be complicated by coagulation disorders (Fig. 32.2).
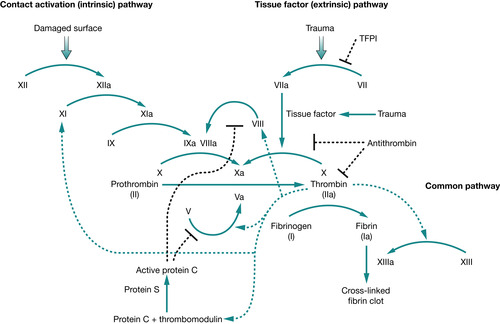 |
| Fig. 32.2 Coagulation cascade |
Interpretation of blood results
Full blood count
The full blood count (FBC) is a broad screening test looking at bone marrow cell production which screens for red cell, white cell and platelet abnormalities. FBC is a relatively easy test to perform, but normal ranges will vary from hospital to hospital. FBC results should be interpreted with knowledge of the normal range of the hospital as well as the child’s age, sex and ethnicity as these will affect the results, e.g. the haemoglobin varies between the sexes and with age.
Bone marrow aspiration
Bone marrow aspiration (where the cells of the bone marrow are aspirated in a liquid form into a syringe) and bone marrow trephine (where a core of bone marrow is removed through a large bore needle) are tests to examine the cellular content (aspirate) and the structure (trephine) of the bone marrow. In children these are most commonly performed under general anaesthesia from the posterior iliac crest (Sepion 1990) as a diagnostic test when bone marrow failure, leukaemia, metabolic or immunological conditions are suspected. The bone marrow aspirate is spread onto a slide and is easily examined using a microscope; diseases affecting the marrow can be easily diagnosed.
Clotting screen
The clotting screen is a screen of the three aspects of coagulation described above. The activated partial prothrombin test (APTT) is a test of the intrinsic pathway; the prothrombin time (PT) is a test of the extrinsic pathway, whilst the thrombin time (TT) tests the conversion of fibrinogen to fibrin in the common pathway. If the clotting screen suggest that a coagulation disorder may be the cause of bruising/bleeding further investigations are required. Interpretation of an abnormality in the initial coagulation screen and further tests required are detailed in Table 32.2. Not all haematology laboratories will be able to perform all of these tests; referral to a regional paediatric haemophilia centre should be considered if an inherited coagulation disorder is suspected.
| PT = prothrombin time; APTT = activated partial thromboplastin time; TT = thrombin time; N = within normal range; ↑ = prolonged; vWD = von Willebrand’s disease; DRVVT = dilute Russell Viper venom time; INR = international ratio. | ||||
| PT | APTT | TT | Possible abnormality | Further investigation required |
|---|---|---|---|---|
| ↑ | N | N | Factor VII deficiency (extrinsic pathway) Liver disease Vitamin K deficiency | Measurement of PT based factors |
| N | ↑ | N | Deficiency of factor VIII (haemophilia or vWD) factor IX, XI, XII or contact factors (intrinsic pathway) Lupus anticoagulant or other coagulation factor inhibitor | Measurement of APTT based factors vWD screen DRVVT, Exner |
| N | N | ↑ | Hypofibrinogenaemia Dysfibrinogenaemia | Reptilase time and other thrombin time corrections |
| ↑ | ↑ | N | Deficiency of factor II, V, X (common pathway) Vitamin K deficiency Liver disease Massive transfusion Oral anticoagulants | PT and APTT based factors, INR |
| N | ↑ | ↑ | Heparin | Reptilase time and other thrombin time corrections |
| ↑ | ↑ | ↑ | Disseminated intravascular coagulation Large amounts heparin Severe hypofibrinogenaemia | D-dimers, reptilase time and other thrombin time corrections |
| N | N | N | All tests normal but history of bleeding – consider: • Factor XIII deficiency • Mild platelet disorders | vWD screen Factor XIII screen/activity Platelet function tests |
Non-malignant haematological conditions
Non-malignant haematological conditions lead to a variety of disorders, which present throughout childhood. The majority of these conditions are inherited, e.g. sickle cell anaemia and haemophilia, and usually present in infancy and early childhood. Acquired diseases, which may present in later childhood and adolescence are often severe and may be life threatening causing problems of infection and bleeding. The commonest non-malignant disorders seen in childhood are discussed below; further information on these disorders can be obtained from the paediatric haematology reference books listed in the bibliography.
Bone marrow failure syndromes
Bone marrow failure can be either congenital or acquired. Although extremely rare the commonest form of congenital bone marrow failure is Fanconi anaemia (FA), which is inherited in an autosomal manner and is associated with growth retardation, defects of the skeleton, kidneys or skin, with occasional mental retardation (Hoffbrand et al 2001 p 91). Children with FA usually present at around 5–10 years of age with increasing signs of bone marrow failure that may include pancytopenia (reduction in blood count of red and white blood cells and platelets) or leukaemia.
Aplastic anaemia (AA) also presents with pancytopenia which may be either congenital or acquired. Causes of acquired AA include infection, drugs (especially chemotherapy) and exposure to radiation or pesticides. Kostmann syndrome is rare, also known as severe congenital neutropenia (SCN), and usually detected soon after birth when severe and/or life-threatening infection occurs (Ancliff 2003).
Osteopetrosis (literally ‘stone bone’) is a rare inherited disorder where bones harden becoming brittle. Mild osteopetrosis is usually asymptomatic but severe forms result in stunted growth, deformity, anaemia from bone marrow failure, blindness and deafness due to increased pressure on nerves from bone overgrowth (Hamdan et al 2006).
Haemoglobinopathy – in infancy children have a variant of haemoglobin known as fetal haemoglobin or HbF. After about 6 months of age they begin to express adult haemoglobin (HbA). Children inherit two sets of haemoglobin genes, one set from each parent. In haemoglobinopathies there is an abnormality in one or both sets of the haemoglobin genes giving rise to an abnormal haemoglobin molecule, which leads to diseases known collectively as haemoglobinopathies.
One of the most significant genetic abnormalities in haemoglobin causes sickle cell anaemia, which is the most common genetic defect in England, affecting 1:2000 births (Dick 2007) this is most commonly seen in people of Afro-Caribbean decent. The abnormal haemoglobin (HbS) is fragile and unable to carry oxygen effectively, the red cells ‘sickle’ and breakdown causing symptoms such as pain, anaemia and stroke. Children with only one copy of this gene are said to have sickle cell trait (HbAS). Sickle cell disease occurs when two copies of the HbS gene are inherited (HbSS) – one from each parent. See Table 32.3 for a list of haemoglobin gene variants.
| Haemoglobin variant | Clinical features | |
|---|---|---|
| Fetal haemoglobin (HbF) | Normal feature of infancy Switch to adult Hb (HbA) at 6 months of age | Hereditary persistence of HbF caused by genetic abnormalities – leads to anaemia. |
| Haemoglobin A (HbA) | Normal adult haemoglobin | None |
| Haemoglobin C (HbC) | Most frequently seen in children of west African decent | Mild haemolytic anaemia with splenomegaly |
| Haemoglobin D (HbD) | No haematological abnormality to mild haemolytic anaemia | |
| Haemoglobin E (HbE) | Most frequently seen in children of south east Asian decent | Mild hypochromic anaemia |
| Haemoglobin S (HbS) | Sickle haemoglobin | Sickle cell trait (HbAS) – not usually clinically significant Additional care with anaesthesia Sickle cell disease (HbSS) – severe haemolytic anaemia with sickle cell crises including stroke in young children |
| Haemoglobin SC (HbSC) | Particular risk of thrombosis and pulmonary embolism | |
| α thalassaemia | Most commonly seen in children from Mediterranean/Asian/Oriental descent | Can be mild causing moderately severe hypochromic anaemia but in severe cases is incompatible with life, can cause hydrops fetalis and intra uterine death |
| β thalassaemia | Most commonly seen in children from Mediterranean/Asian/Oriental decent | Severe anaemia from 6 months of age requiring transfusion Hepatosplenomegaly Infections Osteoporosis |
Thalassaemia is a group of disorders of the globin genes where one or more of the genes is either missing or is inactive, this is most commonly seen in children of Mediterranean and Asian decent (Modell et al 2001). There are two subtypes of thalassaemia: α (alpha) and β (beta) thalassaemia. The α subtype causes moderately severe anaemia and splenomegaly unless two copies of the abnormal α-thalassaemia haemoglobin are inherited which results in incompatibility with life often resulting in inter uterine death with hydrops fetalis (Yang & Li 2009). The β subtype is a much more clinically severe disease presenting in infancy with anaemia, hepatosplenomegaly, bone marrow hyperplasia, infection and osteoporosis (Clarke & Higgins 2000).
There is now an national newborn haemoglobinopathy screening programme, with all neonates in the UK undergoing haemoglobin analysis as part of the neonatal screening programme (previously known as the Guthrie test) (NHS 2006). This has resulted in ‘at risk’ children being diagnosed at an average of 6 weeks before they become symptomatic (Bain 2009).
Bleeding disorders
The disorders which result in bleeding can be mild to severe, inherited or acquired, as a result of bone marrow abnormalities in platelet production or function or in a reduction or absence of any of the coagulation factors. The commonest causes of bleeding are discussed below as either disorders of platelets or coagulation.
Platelet disorders
Thrombocytopenia
The full blood count will show if there is thrombocytopenia; this should be repeated to confirm this finding. The commonest cause of thrombocytopenia in children is immune thrombocytopenic purpura (ITP), which is often occurs following viral infections (Vora & Makrish 2001). Whilst bruising is common severe bleeding is rare if the platelet count is above 20 × 10 9/L. If there is persistent thrombocytopenia, referral to a paediatric haematology centre should be considered, aiming to exclude a congenital platelet disorder, leukaemia or a bone marrow failure syndrome. Congenital thrombocytopenias are rare but may present with symptoms of bruising or bleeding soon after birth. Thrombocytopenia with absent radii (‘TAR syndrome’) is an autosomal recessive disorder characterised by bilateral absence of the radii, which is clinically obvious at birth (Al-Jefri et al 2000). Wiskott–Aldrich syndrome is an X-linked immune deficiency disorder associated with bacterial infections and/or eczema (Mullen et al 1993). Bernard–Soulier syndrome is an autosomal recessive disorder most commonly seen in consanguineous families which causes mild to severe bleeding in both boys and girls (George et al 1981).
Functional
These are platelet disorders with normal platelet numbers but abnormal function and can be minor causing bleeding following surgery, through to life threatening such as Glanzmann’s thrombasthenia. Transient abnormalities in platelet function causing easy bruising are common and are often associated with the use of anti-platelet drugs such as non-steroidal anti-inflammatory drugs or aspirin. Other drugs that are known to affect platelet function are listed in Table 32.4.
| Cytotoxic therapy |
| Ethanol |
| Chloramphenicol |
| Arsenic |
| Benzene |
| Non-steroidal anti-inflammatory drugs |
| Aspirin |
| Rifampicin |
| Penicillin |
| Sulphonamides |
| Trimethoprim |
| Diazepam |
| Sodium valproate |
| Carbamazepine |
| Frusemide |
| Tolbutamide |
| Digoxin |
| Heparin |
| Warfarin |
| Methyldopa |
| Oxyprenolol |
| Quinine |
Inherited platelet function defects are very rare, the most severe of these is Glanzmann’s thrombasthenia – an autosomal recessive disorder which results in severe often spontaneous bleeding usually from the mucous membranes, which can be life threatening (Hardisty 2000). Platelet storage pool disorders, where there is a deficiency in the nucleotide content of the platelet, and platelet release defects, where the nucleotides cannot be released properly, both result in mild to moderate bruising and bleeding following trauma or surgery.
Coagulation disorders
Von Willebrand disease (vWD) is the commonest inherited bleeding disorder with an incidence of 1:100 to 1:1000. The inheritance varies according to the subtype and affects boys and girls equally. There are three major subtypes of vWD; bleeding in type 1 and 2 is usually mild but in type 3, which is inherited in an autosomal recessive manner, there is usually severe bleeding often from mucous membranes from early childhood resulting in mouth and nose bleeds (Mannucci 2001).
Haemophilia is an X-linked condition in which boys experience bruising and bleeding which can be severe (Khair et al 2003). Haemophilia A (classical haemophilia, a deficiency of factor VIII) affects about 1:10,000 boys, whilst haemophilia B (Christmas disease or factor IX deficiency) affects about 1:50,000 boys. The bruising/bleeding is dependent upon the level of factor in the plasma, with the severest bleeding occurring in those with the lowest factor levels. Although considered an inherited disorder, approximately 1 in 3 boys with haemophilia have no previous family history with there being a new genetic mutation in their FVIII or FIX gene. Girls who are haemophilia carriers may also suffer bruising/bleeding and mennorhagia when in adolescence as they may also have low factor levels.
 Scenario 1
Scenario 1
Scenario 1Jennifer is a 4-year-old girl who has sickle cell disease. She has been admitted to the ward from the A&E department with acute chest syndrome and is accompanied by her mum. Jennifer has been ‘off colour’ for the last 4 days with fever and cough, her brother has similar symptoms though not sickle cell disease.
• What framework would you use to assess Jennifer?
• What observations do you need to do?
• What do you expect these observations to be?
• Is Jennifer’s condition mild, moderate or severe?
• Is there anything in the history that would give concern?
• What care would you expect to be initiated?
• What ongoing care does Jennifer need?
The other coagulation disorders are very rare compared with haemophilia and vWD; however they can cause life-threatening bleeding. Deficiency of factor II, V, VII or X can cause significant bleeding when autosomal recessive inheritance has occurred, however carriers of these disorders can also experience mild bleeding/bruising (Bolton-Maggs et al 1995). Factor XI deficiency is most commonly seen in children of Ashkenazi Jewish decent; the bleeding tendency is usually mild (Collins et al 1995). Factor XII deficiency is commonly seen but rarely if ever predisposes to bruising/bleeding. Factor XIII deficiency is an autosomal recessive condition which presents in the neonatal period with umbilical cord bleeding, delayed cord separation or intracranial haemorrhage (Anwar & Miloszeski 1999). Disorders of fibrinogen can be both in quantity and/or quality, often presenting in early childhood with bruising and bleeding whilst dysfibrinogenaemia may also cause thrombosis (Lak et al 1999).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Get Clinical Tree app for offline access
