17 Neurological Alterations and Management
After reading this chapter, you should be able to:
• differentiate cerebral hypoxia from cerebral ischaemia and focal from global ischaemia
• differentiate between primary and secondary brain injuries due to brain injury
• relate the procedures of selected neurodiagnostic tests to nursing implications for patient care
• discuss the rationale for medical and nursing management in the care of the brain-injured patient.
Concepts of Neurological Dysfunction
Alterations in Consciousness
Altered Cognition and Coma
Coma is a state of unresponsiveness from which the patient, who appears to be asleep, cannot be aroused by verbal and physical stimuli to produce any meaningful response; therefore, the diagnosis of coma implies the absence of both arousal and content of consciousness.1 Coma must be considered a symptom with numerous causes, different natural modes, and several management modes.
Somnolence is a state of unconsciousness from which the patient can be fully awakened. Although there are many specific causes of unconsciousness, the sites of cerebral affection are either the bilateral cerebral cortex or the brainstem reticular activating system. The commonest causes of bilateral cortical disease are deficiencies of oxygen, metabolic disorders, physical injury, toxins, postconvulsive coma and infections.2 The reticular activating system maintains the state of wakefulness through continuous stimulation of the cortex. Any interruption may lead to unconsciousness. The reticular activating system can be affected in three principal ways: by supratentorial pressure, by infratentorial pressure, and by intrinsic brainstem lesions. Supratentorial lesions produce impaired consciousness by enlarging and displacing tissue. Lesions that affect the brainstem itself damage the reticular activating system directly.
Aetiology of altered cognition
Recently gained confusion, severe apathy, stupor or coma implies dysfunction of the cerebral hemispheres, the diencephalon and/or the upper brainstem.3 Focal lesions in supratentorial structures may damage both hemispheres, or may produce swelling that compresses the diencephalic activating system and midbrain, causing transtentorial herniation and brainstem damage. Primary subtentorial (brainstem or cerebellar) lesions may compress or directly damage the reticular formation anywhere between the level of the midpons and, (by upward pressure), the diencephalon. Metabolic or infectious diseases may depress brain functions by a change in blood composition or the presence of a direct toxin. Impaired consciousness may also be due to reduced blood flow (as in syncope or severe heart failure) or a change in the brain’s electrical activity (as in epilepsy). Concussion, anxiolytic drugs and anaesthetics impair consciousness without producing detectable structural changes in the brain.
Many of the enzymatic reactions of neurons, glial cells, and specialised cerebral capillary endothelium in the brain must be catalysed by the energy-yielding hydrolysis of adenosine triphosphate (ATP) to adenosine diphosphate (ADP) and inorganic phosphate. Without a constant and generous supply of ATP, cellular synthesis slows or stops, neuronal functions decline or cease, and cell structures quickly fall apart.4 The brain depends entirely on the process of glycolysis and respiration within its own cells to provide its energy needs. Even a short interruption of blood flow or oxygen supply threatens tissue vitality.
Seizures
A seizure is an uninhibited, abrupt discharge of ions from a group of neurons resulting in epileptic activity.5 The majority of patients experiencing seizures in the ICU do not have preexisting epilepsy, and their chances of developing epilepsy in the future are usually more dependent on the cause than on the number or intensity of seizures that they experience. However, because of other deleterious neuronal and systemic effects of seizures, their rapid diagnosis and suppression during a period of critical illness is necessary.
Aetiology of seizures
Seizures may either prompt the patient’s admission to ICU (because of status epilepticus) or develop as a complication of another illness.6 Seizures can be due to vascular, infectious, neoplastic, traumatic, degenerative, metabolic, toxic or idiopathic causes. Factors influencing the development of posttraumatic epilepsy include an early posttraumatic seizure, depressed skull fracture, intracranial haematoma, dural penetration, focal neurological deficit and posttraumatic amnesia (PTA) over 24 hours with the presence of a skull fracture or haematoma. Seizures in critically ill patients are most commonly due to drug effects; metabolic, infectious or toxic disorders; and intracranial mass lesions although they may be due to trauma or neoplasm.7 Conditions producing seizures tend either to increase neuronal excitation or to impair neuronal inhibition. A few generalised disorders (e.g. non-ketotic hyperglycaemia) may produce partial or focal seizures.
Alterations in Motor and Sensory Function
Muscle tone, which is a necessary component of muscle movement, is a function of the muscle spindle (myotatic) system and the extrapyramidal system, which monitors and buffers input to the lower motor neurons by way of the multisynaptic pathways.8 Upper motor neuron lesions produce spastic paralysis, and lower motor neuron lesions produce flaccid paralysis. Damage to the upper motor and sensory neurons of the corticospinal, corticobulbar and spinothalamic tracts is a common component of stroke.9 Polyneuropathies involve multiple peripheral nerves and produce symmetrical sensory, motor, and mixed sensorimotor deficits:
• Lesions of the corticospinal and corticobulbar tracts: result in weakness or total paralysis of predominantly distal voluntary movement, Babinski’s sign (i.e. dorsiflexion of the big toe and fanning of the other toes in response to stroking the outer border of the foot from heel to toe), and often spasticity (increased muscle tone and exaggerated deep tendon reflexes).
• Disorders of the basal ganglia: (extrapyramidal disorders) do not cause weakness or reflex changes. Their hallmark is involuntary movement (dyskinesia), causing increased movement (hyperkinesias) or decreased movement (hypokinesia) and changes in muscle tone and posture.
• Cerebellar disorders: cause abnormalities in the range, rate and force of movement. Strength is minimally affected.
Autonomic Nerve Dysfunction
There is strong evidence for numerous interactions among the central nervous system (CNS), peripheral nervous system (both sympathetic and parasympathetic branches), the endocrine system, and the immune system, hence ANS dysfunction is related to that complex triad.10 Autonomic nerve (AN) dysfunction ranges from alterations in the sympathetic–parasympathetic balance to almost complete cessation as occurs in spinal cord injury. As the ANS controls organ function AN dysfunction is related to all-organ alteration and failure. The immune system is connected to the nervous system through the ANS with many of the patients with infections, systemic inflammatory response and multi-organ failure exhibiting AN dysfunction. AN dysfunction is closely related to systemic inflammation hence those with conditions with increased levels of inflammatory markers such as chronic disease and obesity have predisposing AN dysfunction. AN dysfunction is assessed by time and spectral domain heart rate variability and is currently being researched as a neurological assessment technique.11
Alterations in Cerebral Metabolism and Perfusion
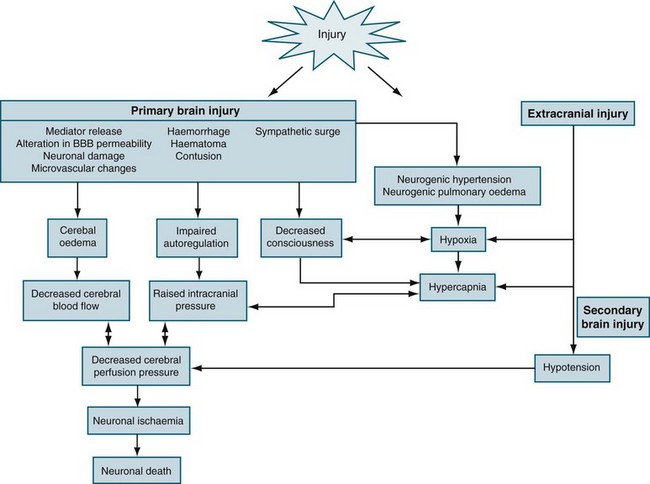
For decades, impairment of cerebral metabolism has been attributed to impaired oxygen delivery, mediated by reduced cerebral perfusion in the swollen cerebral parenchyma. Accordingly, reduction of ICP is usually argued for restoration of previously compromised cerebral perfusion for improvement of cerebral metabolism. Although uncontrolled ICP elevation has been shown to be responsible for reduced oxygen delivery, non-ischaemic impairment of oxidative metabolism and mitochondrial damage has only recently been recognised as a prominent source of energy crisis triggered by brain injury in the presence of adequate cerebral blood flow.12 Accumulating evidence has shown that the mitochondrion has a pivotal role in post traumatic neuronal death by integrating numerous noxious signals responsible for both structural and functional damage on one hand and by amplifying these signals through activation of several cellular signalling events leading to cell death. In addition, more complex processes with the alteration of cerebral perfusion, such as cerebral hypoperfusion, ischaemia, reperfusion injury, inflammation and oedema result in increased intracranial pressure (ICP).
Cerebral Ischaemia
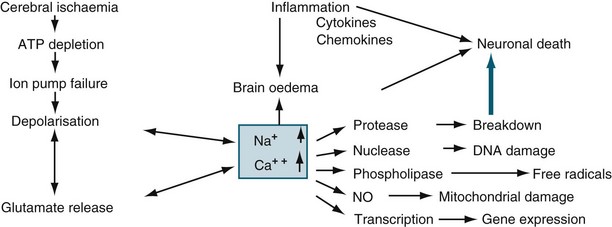
Ischaemia is the inadequate delivery of oxygen, the inadequate removal of carbon dioxide from the cell, and an increase in the production of intracellular lactic acid. Ischaemia can be caused by an increase in nutrient utilisation by the brain in a hyperactive state, a decrease in delivery related to either cerebral or systemic complications, and/or a mismatch between delivery and demand.13 The ischaemic cascade is described in Figure 17.1. Inflammation, together with oxidative stress, excitotoxicity, disrupted calcium homeostasis and energy failure, is one of the key pathological changes in ischaemic brain damage.14 There is a significant inflammatory response in ischaemic brains, including leucocyte and monocyte infiltration into the brain, activation of microglia and astrocytes, elevated production of inflammatory cytokines and chemokines and increased expression and activity of adhesion molecules, complement and metalloproteinases. Of importance, brain ischaemia can lead to significant inflammatory responses in the central nervous system and can also cause significant changes in the peripheral immune system. There are two phases. In the relatively early phase, activated spleen cells and lymph nodes and blood mononuclear cells secrete significantly enhanced levels of TNF-α, IL-6 and IL-2. This then results in global immunosuppression affecting the spleen, lymph nodes, thymus and a significant decrease in the number of immune cells in the circulation.15 When cerebral blood flow (CBF) falls to about 40% of normal, EEG slowing occurs. When CBF falls below 10 mL/100 g/min (20%), the function of ionic pumps fails, which leads to membrane depolarisation. Cerebral ischaemia and reperfusion injury contribute to the cascade of physiological events, termed secondary brain injury. Recent studies have shown that low-dose paracetamol reduces inflammatory protein release from brain endothelial cells exposed to oxidant stress16 and that propofol protects against neuronal apotosis.17
Cerebral Oedema
Cerebral oedema is defined as increased brain water content. The brain is particularly susceptible to injury from oedema, because it is located within a confined space and cannot expand, and because there are no lymphatic pathways within the CNS to carry away the fluid that accumulates. The white matter is usually much more involved, as myelinated fibres have a loose extracellular space, while the grey matter has a much higher cell density with many connections and much less loose extracellular space.18 The two main subdivisions of cerebral oedema are extracellular and intracellular.
Intracellular (cytotoxic) oedema
Cellular swelling, usually of astrocytes in the grey matter, is generally seen after cerebral ischaemia caused by cardiac arrest or minor head injury.19 The blood–brain barrier (BBB) is intact and capillary permeability is not impaired. The cause of intracellular oedema is anoxia and ischaemia; it is usually not clinically significant, and is reversible in its early phases.
Extracellular (vasogenic) oedema
Extracellular oedema involves increased capillary permeability, and had been termed ‘BBB breakdown’.20 Rises in brain water content with extracellular oedema are often quite dramatic, because the fluid that results from increased capillary permeability is usually rich in proteins, resulting in the spread of oedema and brain ischaemia. This can lead to cytotoxic oedema, and to the progressive breakdown of both astrocytes and neurons.19 While the classification of oedema is useful to define specific treatments, it is somewhat arbitrary, as cytotoxic and vasogenic oedema often occur concurrently. In fact, each of these processes may cause the other. Ultimately, these changes can lead to raised intracranial pressure and herniation.
Hydrocephalus
Hydrocephalus is the result of an imbalance between the formation and drainage of cerebrospinal fluid (CSF). Reduced absorption most often occurs when one or more passages connecting the ventricles become blocked, preventing movement of CSF to its drainage sites in the subarachnoid space just inside the skull.21 This type of hydrocephalus is called ‘non-communicating’. Reduction in absorption rate, called ‘communicating hydrocephalus’ can be caused by damage to the absorptive tissue. Both types lead to an elevation of the CSF pressure within the brain. A third type of hydrocephalus, ‘normal pressure hydrocephalus’, is marked by ventricle enlargement without an apparent rise in CSF pressure, which mainly affects the elderly.
Hydrocephalus may be caused by: congenital brain defects; haemorrhage, in either the ventricles or the subarachnoid space; CNS infection (syphilis, herpes, meningitis, encephalitis or mumps); and tumours. Irritability is the commonest sign of hydrocephalus in infants and, if untreated, may lead to lethargy. Bulging of the fontanelle, the soft spot between the skull bones, may also be an early sign. Hydrocephalus in infants prevents fusion of the skull bones, and causes expansion of the skull. Symptoms of normal pressure hydrocephalus include dementia, gait abnormalities and incontinence.22 Treatment includes ventriculostomy drainage of CSF in the short term, or a surgical shunt for those with chronic conditions. Either is predisposed to blockage and infection.
Intracranial Hypertension
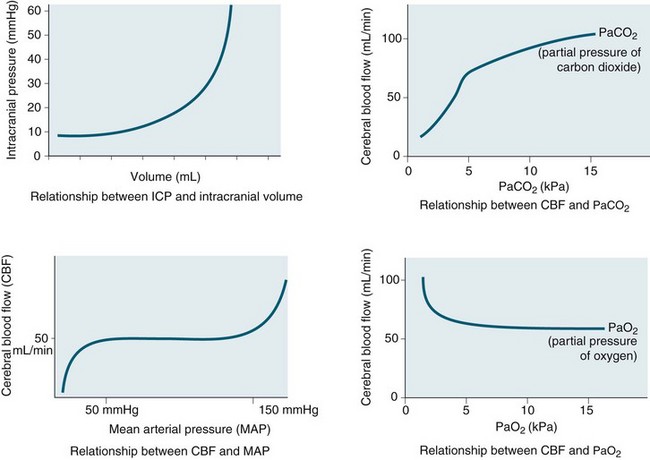
Intracranial pressure is the pressure exerted by the contents of the brain within the confines of the skull and the BBB. The Munro–Kelly hypothesis states that the contents of the cranium (60% water, 40% solid) are not compressible and thus an increase in volume causes a rapid rise in pressure and changes to the compensatory reserve and pulse amplitude, as illustrated in Figure 17.2.23 Normal ICP is 0–10 mmHg, and a sustained pressure of >15 mmHg is termed intracranial hypertension, with implications for CBF.24 Areas of focal ischaemia appear when ICP is >20 mmHg and global ischaemia occurs at >50 mmHg. ICP waveform contains valuable information about the nature of cerebrospinal pathophysiology. ICP increased to the level of systemic arterial pressure extinguishes cerebral circulation, which will restart only if arterial pressure rises sufficiently beyond the ICP to restore cerebral blood flow. Autoregulation of cerebral blood flow and compliance of the cerebrospinal system are both expressed in ICP. Methods of waveform analysis are useful, both to derive this information and to guide the management of patients.25
Initially, intracranial compliance allows compensation for rises in intracranial volume due to autoregulation. During a slow rise in volume in a continuous mode, the ICP rises to a plateau level at which the increased level of CSF absorption keeps pace with the rise in volume with ample compensatory reserve. This is expressed as an index, as shown in Figure 17.3.26 Intermittent expansion causes only a transient rise in ICP at first. When sufficient CSF has been absorbed to accommodate the volume, the ICP returns to normal. The ICP finally rises to the level of arterial pressure which itself begins to rise, accompanied by bradycardia or other disturbances of heart rhythm (termed the Cushing’s response). This is accompanied by dilation of the small pial arteries and some slowing of venous flow, which is followed by pulsatile venous flow.
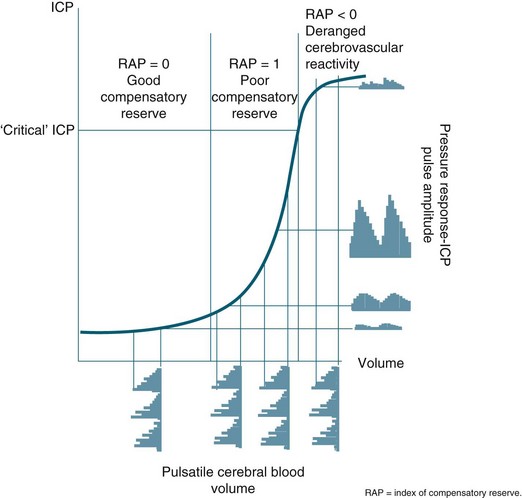
FIGURE 17.3 In a simple model, pulse amplitude of intracranial pressure (ICP) (expressed along the y-axis on the right side of the panel) results from pulsatile changes in cerebral blood volume (expressed along the x-axis) transformed by the pressure–volume curve. This curve has three zones: a flat zone, expressing good compensatory reserve, an exponential zone, depicting poor compensatory reserve, and a flat zone again, seen at very high ICP (above the ‘critical’ ICP) depicting derangement of normal cerebrovascular responses. The pulse amplitude of ICP is low and does not depend on mean ICP in the first zone. The pulse amplitude increases linearly with mean ICP in the zone of poor compensatory reserve. In the third zone, the pulse amplitude starts to decrease with rising ICP. RAP, index of compensatory reserve.26
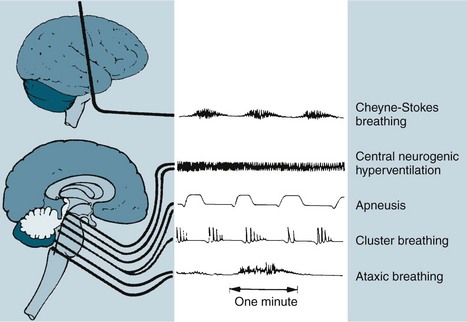
The respiratory changes depend on the level of brainstem involved. A midbrain involvement results in Cheyne-Stokes respiration. When the midbrain and pons are involved, there is sustained hyperventilation. There are rapid and shallow respirations with upper medulla involvement, with ataxic breathing in the final stages (see Figure 17.4).27
Often, neurogenic pulmonary oedema may occur due to increased sympathetic activity as a result of the effects of elevated ICP on the hypothalamus, medulla or cervical spinal cord. The causes of intracranial hypertension are classified as acute or chronic. Acute causes include brain trauma, ischaemic injury and intracerebral haemorrhage. Infections such as encephalitis or meningitis may also lead to intracranial hypertension. Chronic causes include many intracranial tumours, such as ependymomas, or subdural bleeding that may gradually impinge on CSF pathways and interfere with CSF outflow and circulation. As the ICP continues to increase, the brain tissue becomes distorted, leading to herniation and additional vascular injury.28
Neurological Therapeutic Management
Optimising Cerebral Perfusion and Oxygenation
Intracranial hypertension and cerebral ischaemia are the two most important secondary injury processes that can be anticipated, monitored and treated in the ICU. This applies to all aetiologies of brain injury including trauma. This section discusses the modalities of neuroprotection, including the management of intracranial hypertension, vasospasm and cerebral ischaemia. Nursing interventions for the prevention of secondary insults and promotion of cerebral perfusion are described in Table 17.1. Importantly, the aims of nursing management are based on published guidelines and are directed at optimising cerebral perfusion and metabolism by various initiatives.
TABLE 17.1 Nursing interventions for the promotion of cerebral perfusion in acute brain injury
Management of Cerebral Oxygenation and Perfusion
Cerebral monitoring in brain-injured patients has focused on the prevention of secondary injury to the brain owing to impaired perfusion. However, ICP monitoring and ICP manipulation does not equal cerebral oxygenation.29 There are currently four techniques that can be used to assess cerebral oxygenation: jugular venous oxygen saturation, positron emission tomography, near-infrared spectroscopy, and brain tissue oxygenation monitoring (PbtO2). Their strengths and weaknesses are the subject of several recent reviews.30,31 The selection among these forms of oxygenation monitoring is focused on the appropriateness of focal or global monitoring, the location of the monitor in relation to the injury, and the intermittent or continuous nature of the monitoring. The use of PbtO2, as assessed by the intraparenchymal polarographic oxygen probe, has the advantage of directly monitoring the zone of injury and thus earlier detection of perfusion abnormalities that may impact global cerebral oxygenation later. This may also allow the rescue of watershed areas of perfusion. However, there is controversy regarding the appropriate placement of such monitors. Insertion of the probe into non injured areas yields data equivalent to global assessments of cerebral oxygenation. Consequently, close attention should be paid to the location of the catheter in relation to the injury in interpretation and use of PbtO2 results. Jugular venous oxygen saturation (SjO2) is representative of global cerebral oxygen metabolism, but technically it is difficult to obtain reproducible results. Cerebral tissue oxygenation values of <20 mmHg are targeted for intervention based on Brain Trauma Foundation (BTF) guidelines but only at level III evidence.32 PbtO2 can be increased by increasing the FiO2/PaO2 ratio and by reducing cerebral metabolic requirements for oxygen (CMRO2) using brain temperature control with active cooling and metabolic rate control with sedation and adequate feeding. Additional interventions such as volume infusion, transfusion, and inotropic support directed at improving cardiac output can also be used to increase oxygen delivery.33
Brain inflammation after injury contributes to impaired oxygenation and perfusion, but currently its management has not translated to successful clinical management. However, the use of cerebral microdialysis (MD) and the measurement of biochemical markers (lactate, glutamate, pyruvate, glycerol and glucose) of cerebral inflammation and metabolism do contribute towards early warnings of impending hypoxia/ischaemia and neurological deterioration, and this may allow timely implementation of neuroprotective strategies. Elevation of the lactate/pyruvate ratio is typically seen in cerebral ischaemia and mitochondrial dysfunction, and has been used to tailor therapy.34 However, MD reflects only local tissue biochemistry and the accurate placement of the catheter is crucial. Furthermore, because there are wide variations in measured variables, trend data are more important than absolute values. Although MD is used routinely in a few centres it has not yet been introduced into widespread clinical practice and, at present, should be considered a research tool for use in specialist centres. MD has the potential to become established as a key component of multi-modality monitoring during management of acute brain injury during neurointensive care.
Management of Intracranial Hypertension
1. CSF by ventricular drainage (as discussed previously)
2. cerebral blood volume by hyperventilation, osmotic diuretic therapy or hypothermia
3. brain tissue water content by osmotic diuretic therapy
4. removing swollen and irreversibly injured brain
Hyperventilation
Hyperventilation reduces PaCO2 and will reduce ICP by vasoconstriction induced by alkalosis but it also decreases cerebral blood flow.35 The fall in ICP parallels the fall in CBV. Hyperventilation decreases regional blood flow to hypoperfused areas of the brain. Thus, generally PaCO2 should be maintained in the low normal range of about 35 mmHg. Hyperventilation should be utilised only when ICP elevations are refractory to other methods and when brain tissue oxygenation is in the normal range.36 The BTF Guidelines recommend hyperventilation therapy only for brief periods when there is no neurological deterioration or for longer periods when ICP is refractory to other therapies.32
Osmotherapy
Acute administration of an osmotic such as mannitol or hypertonic saline produces a potent antioedema action, primarily on undamaged brain regions with an intact BBB. This treatment causes the movement of water from the interstitial and extracellular space into the intravascular compartment, thereby improving intracranial compliance or elastance. In addition to causing ‘dehydration’ of the brain, osmotic agents have been shown to exert beneficial non-osmotic cerebral effects, such as augmentation of cerebral blood flow (by reducing blood viscosity, resulting in enhanced oxygen delivery), free radical scavenging, and diminishing CSF formation and enhancing CSF reabsorption.37
The BTF recommends mannitol in intracranial hypertension in bolus administration, keeping the serum osmolarities greater than 320 mOsm/L, plasma Na+ <160 mmol/L and avoiding hypovolaemia. Urine output after mannitol administration needs to be replaced, generally with normal saline. Brain free water is increased with 5% dextrose and hyperglycaemia; hence these need to be avoided. The use of frusemide in conjunction with mannitol promotes a synergistic action, particularly in patients refractory to mannitol alone. Recent studies now suggest that mannitol and frusemide have antiepileptic properties38 and that mannitol has a role in ischaemic stroke.
Intravenous hypertonic saline (HTS) increases cerebral perfusion and decreases brain swelling and inflammation more effectively than conventional resuscitation fluids. HTS behaves like 20% mannitol in acute cerebral oedema but maintains haemodynamic status. However, unlike HTS, mannitol induces a diuresis, which is relatively contraindicated in patients with both TBI and hypovolaemia as it may worsen intravascular volume depletion and decrease cerebral perfusion. Therefore, despite theoretical advantages of HTS resuscitation in patients with TBI, an Australian randomised controlled trial39 found no difference in outcome between HTS and other resuscitation fluids in prehospital resuscitation. However, in many Australian and New Zealand intensive care units, HTS is used as a preferred alternative to mannitol in patients with raised ICP.
Normothermia
Hyperthermia occurs in up to 40% of patients with ischaemic stroke and intracerebral haemorrhage and in 40–70% of patients with severe TBI or aneurysmal subarachnoid haemorrhage. Hyperthermia is independently associated with increased morbidity and mortality after ischaemic and haemorrhagic stroke, and in subarachnoid haemorrhage and TBI patients temperature elevation has been linked to raised intracranial pressure. Temperature elevations as small as 1–2°C above normal can aggravate ischaemic neuronal injury and exacerbate brain oedema.40 Mild hypothermia protects numerous tissues from damage during ischaemic insult.41 The use of paracetamol, cooling blankets, ice packs, evaporative cooling and new cooling technologies may be useful in maintaining normothermia. Hyperaemia (increased blood flow) may occur during rewarming, resulting in acute brain swelling and rebound intracranial hypertension.42 In an original study, Marion and colleagues.43 demonstrated a higher mortality rate than in more recent trials,44 possibly due to rapid rewarming and rebound hyperaemia and cerebral oedema.
Maintenance of body temperature at 35°C may be optimal.45 Intracranial pressure falls significantly at brain temperatures below 37°C but no difference was observed at temperatures below 35°C. Cerebral perfusion pressure peaks at 35–36°C and decreases with further falls in temperature.45 At a temperature below 35°C, both oxygen delivery and oxygen consumption decrease. Cardiac output decreases progressively with hypothermia. Therefore, cooling to 35°C may reduce intracranial hypertension and maintain sufficient CPP without associated cardiac dysfunction or oxygen debt.46 As the temperature is lowered from 34°C to 31°C, the volume of IV fluid infusion and inotrope requirements increase substantially and, despite such interventions, mean arterial pressure decreases. At 31°C serum potassium, white blood cell count and platelet counts are diminished.47 Thus, it seems that hypothermia to 35°C may be optimal.
Corticosteroids
Excessive inflammation has been implicated in the progressive neurodegeneration that occurs in multiple neurological diseases, including cerebral ischaemia. The efficacy of glucocorticoids is well established in ameliorating oedema associated with brain tumours and in improving the outcome in subsets of patients with bacterial meningitis. Despite encouraging experimental results, clinical trials of glucocorticoids in ischaemic stroke, intracerebral haemorrhage, aneurysmal subarachnoid haemorrhage and traumatic brain injury have not shown a definite therapeutic effect. Furthermore, the CRASH (corticosteroid randomisation after significant head injury) trial demonstrated an increased risk of death from use of steroids from all causes within two weeks of injury, and was stopped early.48 Consequently, the BTF Guidelines state that the use of steroids is not recommended for TBI.32
Barbiturates and sedatives
The BTF Guidelines state that high-dose barbiturate therapy may be considered in haemodynamically-salvageable severe TBI patients with intracranial hypertension refractory to maximal medical and surgical interventions.49 The utilisation of barbiturates for the prophylactic treatment of ICP has not been indicated. Barbiturates exert cerebral protective and ICP-lowering effects through alteration in vascular tone, suppression of metabolism and inhibition of free radical-mediated lipid peroxidation. Barbiturates may effectively lower cerebral blood flow and regional metabolic demands. The lower metabolic requirements decrease cerebral blood flow and cerebral volume. This results in beneficial effects on ICP and global cerebral perfusion. Barbiturates within the BTF guidelines are now included under the heading of Anaesthetics, Analgesics and Sedatives and these also recommend (Level II) that it is beneficial to minimise painful or noxious stimuli as well as agitation as they may potentially contribute to elevations in ICP. Therefore propofol is recommended for the control of ICP, but does not improve mortality or six-month outcome. High dose propofol should be avoided as it can produce significant morbidity.49
Surgical interventions
The European TBI Guidelines suggest that operative management be considered for large intracerebral lesions within the first four hours of injury. The use of unilateral craniectomy after the evacuation of a mass lesion, such as an acute subdural haematoma or traumatic intracerebral haematoma, is accepted practice. Surgery is also recommended for open compound depressed skull fractures that cause a mass effect.50
Decompressive craniectomy, for refractory intracranial hypertension, has been performed since 1977, with a significant reduction in ICP for both TBI50 and ischaemic stroke.51 In 2011 a multi-centre prospective randomised trial of early decompressive craniectomy in patients with severe traumatic brain injury reported that in adults with severe diffuse traumatic brain injury and refractory intracranial hypertension, early bifrontotemporoparietal decompressive craniectomy decreased intracranial pressure and the length of stay in the ICU but surprisingly was associated with more unfavorable outcomes at both 6 and 12 months using the Extended Glasgow Outcome Scale.52
Prevention of Cerebral Vasospasm
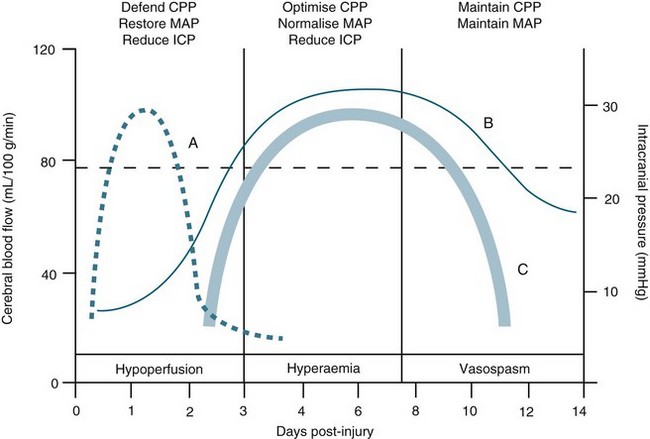
Cerebral vasospasm is a self-limited vasculopathy that develops 4–14 days after subarachnoid haemorrhage (SAH) and/or TBI (see Figure 17.5). Oxyhaemoglobin, a product of haemoglobin breakdown, probably initiates vasoconstriction, leading to smooth-muscle proliferation, collagen remodelling and cellular infiltration of the vessel wall. The resulting vessel narrowing can lead to ischaemia. SAH patients develop cerebral vasospasm, and about one-third develop symptomatic vasospasm, which is associated with neurological signs and symptoms of ischaemia. Posttraumatic brain injury cerebral vasospasm occurs in approximately 10–15% of patients.
Calcium antagonists, such as nimodipine, have not been effective in TBI subarachnoid haemorrhage with vasospasm, and recent studies have suggested that calcium antagonists even prevent neurogenesis after TBI. Nimodipine has demonstrated effectiveness in the treatment of vasospasm in aneurysmal SAH and is now an option for recommended practice. An initial study of nimodipine in patients with TBI demonstrated no difference in outcome, and a Cochrane Systematic Review supports this conclusion.53
Magnesium may prevent cerebral vasospasm through several mechanisms. Increased ATP entry into cells could decrease ischaemic depolarisation and limit infarction size. Magnesium also both inhibits the presynaptic release of excitatory amino acids and is a non-competitive antagonist to postsynaptic NMDA receptors. The drug can also cause vasodilation by inhibiting calcium channel-mediated smooth muscle contraction. Finally, magnesium increases cardiac contractility, which may improve cerebral perfusion in dysautoregulated brain tissue. TBI animal studies have demonstrated promising neuroprotection, but this is still to be confirmed in clinical trials.54 Magnesium, however, does not cross the intact BBB easily, limiting its effect to injury and disease with leaky BBB. A randomised clinical trial of aneurysmal SAH patients receiving magnesium found that IV magnesium infusion reduced the frequency of delayed cerebral ischaemia in patients with aneurysmal SAH and subsequent poor outcome.55
In SAH, more aggressive intravascular volume expansion and induced hypertension are used in conjunction with haemodynamic monitoring. By maintaining haematocrit at 30–33%, a shift in the oxygen dissociation curve is avoided.56 Haemodilutional therapy increases collateral circulation at the site of haemorrhage, while reducing aggregation of erythrocytes where small vessel spasm has occurred. However, there is some emerging physiological data suggesting that normovolaemic hypertension may be the component most likely to increase cerebral blood flow after subarachnoid haemorrhage. In contrast, hypervolaemic haemodilution is associated with increased complications and might also lower the haemoglobin to excessively low levels.56 Also in aneurysmal SAH, endovascular therapies, such as intra-arterial papaverine infusion, are employed. Papaverine acts immediately and increases arterial calibre and cerebral blood flow, but its effects are short-lived. Balloon angioplasty is particularly effective as a durable means of alleviating arterial narrowing and preventing stroke in patients with symptomatic vasospasm after aneurysmal SAH. The timing of endovascular intubation and use of inotropes in patients with cardiac dysfunction are unresolved issues.57
Central Nervous System Disorders
Traumatic Brain Injury
Head injury is a broad classification that includes injury to the scalp, skull or brain. Traumatic brain injury (TBI) is the most serious form of head injury. The range of severity of TBI is broad, from concussion through to post coma unresponsiveness. The Australian age-standardised incidence rate of TBI in 2004/5 was about 150 per 100,000 population. Approximately 90,211 Australians58,59 and 16,000–22,500 New Zealanders60 are hospitalised for TBI every year. Males aged 15–19 years have the highest incidence rates and suffer TBI at a rate almost three times that of women. The very young (0–4 years) and the very old (over 85 years) are also at increased risk.61 Indigenous Australians suffer TBI at almost three times the rate (410 per 100,000) of non-Indigenous Australians.62 It is estimated that 40,000 Australians are living with a disability as a result of TBI.63 Despite definition issues relating to TBI epidemiology, there was an average annual decline of 5% in the TBI rate to 1997/98 but the incidence has increased since then. An Australian and new Zealand epidemiological study64 of TBI (see Research vignette) found that the mean age was 41.6 years; 74.2% were men; 61.4% were due to vehicular trauma, 24.9% were falls in elderly patients, and 57.2% had severe TBI (Glasgow Coma Scale score ≤8). Twelve-month mortality was 26.9% in all patients and 35.1% in patients with severe TBI.
Aetiology
In Australia, motor vehicle-related trauma accounts for about two-thirds of moderate and severe TBI, with falls and assaults being the next most common causes. New Zealand has a higher proportion of recreational injuries compared to vehicle-related trauma. Sporting accidents and falls account for a far greater percentage of mild injuries. Alcohol is associated with up to half of all cases of TBI. In Australia and New Zealand, blunt trauma (falls and vehicle-related), rather than penetrating (stabbing and firearms) or blast, is the predominant mechanism of injury.63 The transfer of energy to the brain tissue actually causes the damage and is a significant determinant in the severity of injury (and routinely noted in ED on admission). In the past 10 years, the introduction of safer car designs, airbags and other road traffic initiatives (e.g. redesigning hazardous intersections, driver education campaigns, random breath testing and reducing speed limits) have decreased the overall number of road fatalities; improvements in retrieval, neurosurgery and intensive care in the past few decades have enabled many people to survive injuries that would previously have been fatal. Research into and prevention of falls and shaken-baby syndromes has had a small impact on incidence reduction.65,66
Pathophysiology of TBI
TBI is a heterogeneous pathophysiological process (see Figure 17.5). The mechanisms of injury forces inflicted on the head in TBI produce a complex mixture of diffuse and focal lesions within the brain.67 Damage resulting from an injury can be immediate (primary) or secondary in nature. Secondary injury results from disordered autoregulation and other pathophysiological changes within the brain in the days immediately after injury. Urgent neurosurgical intervention for intracerebral, subdural or extradural haemorrhages can mitigate the extent of secondary injury. Scalp lesions can bleed profusely and quickly lead to hypovolaemic shock and brain ischaemia. Cerebral oedema, haemorrhage and biochemical response to injury, infection and increased ICP are among the commonest physiological responses that can cause secondary injury. Tissue hypoxia is also of major concern and airway obstruction immediately after injury contributes significantly to secondary injury. Poor cerebral blood flow, as a result of direct (primary) vascular changes or damage, can lead to ischaemic brain tissue, and eventually neuronal cell death.68 Systemic changes in temperature, haemodynamics and pulmonary status can also lead to secondary brain injury (Figure 17.6). In moderate to severe and, occasionally in mild, injury, cerebral blood flow is altered in the initial 2–3 days, followed by a rebound hyperaemic stage (days 4–7) leading to a precarious state (days 8–14) of cerebral vessel unpredictability and vasospasm.64 More than 30% of TBI patient have AN dysfunction characterised by episodes of increased heart rate, respiratory rate, temperature, blood pressure, muscle tone, decorticate or decerebrate posturing, and profuse sweating.70 Lack of insight into these processes and implementing early weaning of supportive therapies can lead to significant secondary insults.
Focal injury
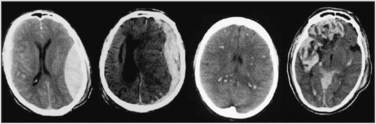
Because of the shape of the inner surface of the skull, focal injuries are most commonly seen in the frontal and temporal lobes, but they can occur anywhere. Contact phenomena are commonly superficial and can generate superficial or contusional haemorrhages through coup and contrecoup mechanisms.71 Cerebral contusions are readily identifiable on CT scans, but may not be evident on day 1 scans, becoming visible only on days 2 or 3. Deep intracerebral haemorrhages can result from either focal or diffuse damage to the arteries.
Diffuse injury
Diffuse (axonal) injury (DAI) refers to the shearing of axons and supporting neuroglia; it may also traumatise blood vessels and can cause petechial haemorrhages, deep intracerebral haematomas and brain swelling.71 DAI results from the shaking, shearing and inertial effects of a traumatic impact. Mechanical damage to small venules as part of the BBB can also trigger the formation of haemorrhagic contusions. This vascular damage may increase neuronal vulnerability, causing post-traumatising perfusion deficits and the extravasation of potentially neurotoxic blood-borne substances. The most consistent effect of diffuse brain damage, even when mild, is the presence of altered consciousness. The depth and duration of coma provide the best guide to the severity of the diffuse damage. The majority of patients with DAI will not have any CT evidence to support the diagnosis. Other clinical markers of DAI include the high speed or force strength of injury, absence of a lucid interval, and prolonged retrograde and anterograde amnesia. Figure 17.7 contrasts CT scans with haematoma formation and DAI.
Mild TBI
Mild TBI often presents as a component of multitrauma or sports injury and can be overlooked at the expense of other peripheral injuries. Risk factors such as vomiting, dizziness, facial and skull fractures; including the loss of CSF from the nose or the ear, will categorise those needing further surveillance. Routine head CT and assessment of PTA are recommended to exclude mass lesions and DAI. Diagnosis and management in the acute phase of mild TBI is as crucial to functional outcome and rehabilitation as in moderate-to-severe TBI.72
Skull fractures
Skull fractures are present on CT scans in about two-thirds of patients after TBI. Skull fractures can be linear, depressed or diastatic, and may involve the cranial vault or skull base. In depressed skull fractures the bone fragment may cause a laceration of the dura mater, resulting in a cerebrospinal fluid leak.73 Basal skull fractures include fractures of the cribriform plate, frontal bones, sphenoid bones, temporal bone and occipital bones. The clinical signs of a basal skull fracture may include: CSF otorrhoea or rhinorrhoea, haemotympanum, postauricular ecchymoses, periorbital ecchymoses, and injury to the cranial nerves: VII (weakness of the face), VIII (loss of hearing), olfactory (loss of smell), optic (vision loss) and VI (double vision).
Nursing Practice
The surveillance and prevention of secondary injury is the key to improving morbidity and mortality outcomes69 (see Table 17.1). It should be noted that in a post hoc in analysis of saline critically ill patients with TBI, fluid resuscitation with albumin was associated with higher mortality rates than was resuscitation with saline.74 Interventions are targeted at maintaining adequate cerebral blood flow and minimising oxygen consumption by the brain in order to prevent ischaemia. The anticipation and prevention of systemic complications are also of vital importance. Assessment is vital to establish priorities in care and is discussed in Chapter 16.
Nursing management of the neurologically impaired, immobilised, mechanically ventilated patient is described in Table 17.2 and is an adaptation of the current guidelines32 (see Table 17.3) to clinical practice (see Online resources for TBI-related protocols). In all TBI multitrauma patients, disability and exposure/environmental control assessment includes the routine trauma series of X-rays, namely chest, pelvis and cervical spine (lateral, anteroposterior and odontoid peg views). These should be reviewed by a radiologist and areas of concern, particularly in the upper and lower regions of the cervical spine, should be clarified with further investigations such as CT scans. Isolated TBI requires CT scanning of the head and upper spine. The management of TBI should include spinal precautions until spinal injury is definitively excluded.
TABLE 17.2 Nursing management of the neurologically impaired, immobilised, mechanically-ventilated patient



