Chapter 8 Neurologic Health Breakdown
When you have completed this chapter you will be able to
WHAT MAKES UP THE NERVOUS SYSTEM: KEY ASPECTS OF ANATOMY AND PHYSIOLOGY
There are two major components of the nervous system: the central nervous system comprising the brain and spinal cord that acts as the control centre for the body, and the peripheral nervous system consisting of the cranial and peripheral nerves. The peripheral nervous system acts as the information pathway, transmitting information between the central nervous system and the sensory receptors, muscles and organs of the body. Through this process, the nervous system organises and coordinates functions of all body systems, as well as consciousness and cognition1,2.
THE NEURONE
The functional unit of the nervous system is the neurone or nerve cell, a specialised cell with the ability to receive and transmit electrochemical nerve impulses. Each neurone is composed of a cell body, an axon or nerve fibre carrying impulses away from the cell body, and processes called dendrites that carry impulses to the cell body (see Figure 8.1). This basic structure is similar for all neurones, but variations occur depending upon the site of the neurone and its specific function. For example, a motor neurone may have a long axon, or fibre, in order to transmit impulses from the central nervous system over a long distance in the body, whereas a sensory nerve may have a shorter axon1,2.
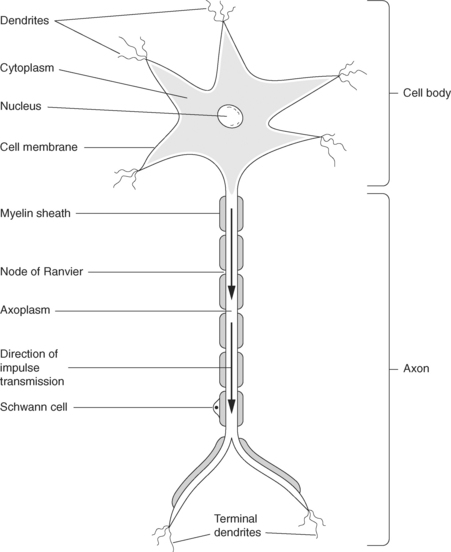
The majority of axons have a fatty (lipoprotein) myelin sheath wrapped around them, with regular constrictions at approximately 1mm intervals called the nodes of Ranvier. Outside the central nervous system, myelin is produced by Schwann cells that lie alongside the nerve, while in the central nervous system, myelin is produced by cells called oligodendrocytes. Unmyelinated nerve fibres are found in the autonomic nervous system, a division of the peripheral nervous system1–3.
NERVE IMPULSE TRANSMISSION
Potassium leaves the cell as it fills with sodium, until the membrane becomes impermeable to sodium ions again and the sodium pump restarts. Three sodium ions then leave the cell, the resting potential of the axon is restored (repolarisation), and two potassium ions passively re-enter the axon. This whole process takes about 2 milliseconds4. A short period of time when another impulse cannot be transmitted (the refractory period) occurs, limiting the frequency of impulses. The transmission of an impulse is an ‘all or nothing’ event, that is, in a healthy neurone the impulse is the same size and speed for a given nerve, and if initiated is always transmitted4. However, the frequency of impulses can vary, with more frequent impulses occurring along the more important pathways of the central nervous system1,4–6.
The speed at which a nerve impulse is transmitted depends on the diameter of the axon, with wider axons transmitting impulses more quickly. If the axon is myelinated, the impulse travels at greater speed, as depolarisation and repolarisation occurs at the nodes of Ranvier as the nerve impulse action potential travels down the axon from one node to the next (saltatory conduction)1–6.
NERVE IMPULSES AND THE SYNAPSE
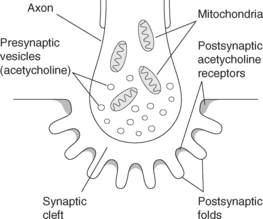
The area where nerves connect with each other, with muscles or other organs is called the synapse. This is where impulses are transmitted from the nerve cell to the target organ through the release of chemical transmitters from the presynaptic neurone to stimulate the postsynaptic neurone/s or organ (see Figure 8.2). There are a number of different chemical transmitters, but the most common one is acetylcholine (ACh). Acetylcholine diffuses across the synaptic cleft and binds to the postsynaptic membrane of a neurone, caus-ing depolarisation of that nerve fibre. In turn, the postsynaptic membrane releases cholinesterase, an enzyme that inactivates the acetylcholine. In the brain, release of neurotransmitter substances is more complex, as is the arrangement of nerve fibres1,4.
CENTRAL NERVOUS SYSTEM
The central nervous system receives, interprets and stores messages from the internal and external environment and transmits messages in return. The brain, a soft mass of tissue, is composed of a collection of nerve cells, white matter and supportive tissue connected to the spinal cord, which has a similar structure. The brain is subdivided into the cerebrum, cerebellum and brainstem (see Figure 8.3).
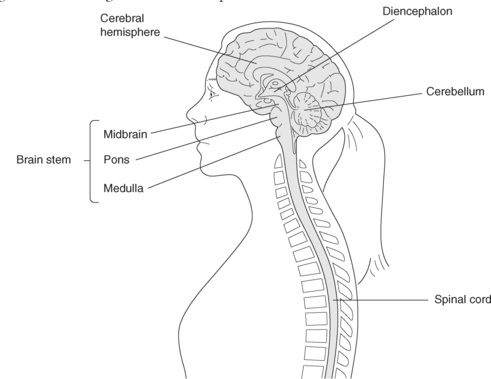
The skull and the vertebrae provide protection for the brain and spinal cord, along with three membranes called the meninges, and cerebrospinal fluid. These three dural membranes, the dura mater, arachnoid mater and pia mater, cover and support the brain. Cerebrospinal fluid (CSF) circulates through the ventricles of the brain, the central canal of the spinal cord, and the subarachnoid space between the arachnoid and pia mater, bathing and cushioning the brain and spinal cord.
CEREBRUM
The cerebrum, the largest part of the brain, consists of a right and left hemisphere, each of which is subdivided into four lobes. The hemispheres are connected by the corpus callosum. The outer layer or cortex of the cerebrum primarily consists of neuronal cell bodies or grey matter, while the inner part of the cerebrum is made up of the axons or white matter. The internal capsule carrying sensory impulses to the cortex, and motor impulses from the cortex to the body, is situated within the white matter in each hemisphere. Sensation and motor function for the left side of the body are controlled by the right cerebrum and on the right side of the body by the left cerebrum, as ascending and descending impulses cross at the medulla oblongata in the brain stem3,4.
The frontal, parietal, temporal and occipital lobes are each responsible for controlling specific bodily functions. The frontal lobes are involved in voluntary movement, speech, intelligence, personality, thought, memory, emotion, and understanding, linking sensory and motor information, and coordinating sensory and motor function. The temporal lobes are responsible for interpretation of hearing, smell and taste, with the dominant temporal lobe responsible for understanding speech. The parietal lobes receive sensory impulses, interpreting information such as shape, size, texture and the sense of body position, and the occipital lobes are the primary area for vision. Cranial nerves 1 and 2 responsible for sight and smell have their origins within the cerebrum1–4.
The hypothalamus, a structure situated at the base of the optic chiasm, acts as an autonomic centre with connections to the brain, spinal cord, autonomic nervous system and the pituitary gland. It is responsible for control of body temperature, appetite, blood pressure, breathing, sleep patterns, behavioural and emotional expression, pituitary secretions, and is partially involved in regulating the stress reaction. The pituitary gland, consisting of an anterior and posterior lobe, is attached to and gets messages from the hypothalamus which regulates production of hormone-stimulating substances that are involved in regulating the functions of other body systems, as well as prolactin, corticotropin, growth hormone, and anti-diuretic hormone1–4.
Within the cerebrum are four ventricles or connected cavities consisting of two lateral ventricles, one in each cerebral hemisphere, the third ventricle, which surrounds the thalamus and the fourth, an expansion of the central canal in the medulla oblongata. About 500mL of cerebrospinal fluid (CSF) is produced in the lateral ventricles every 24 hours. CSF is clear and contains water, glucose, protein and lymphocytes2,3.
CEREBELLUM
The cerebellum, mirrors the structure of the cerebrum having two hemispheres, with an outer cortex of grey matter and an inner core of white nerve fibres, connecting the cerebellum to the cerebrum and brain stem. The cerebellar hemispheres receive sensory input from muscles, joints and the vestibular canals of the inner ear, and control muscle coordination, posture, equilibrium and balance on the same side of the body.
BRAIN STEM
Voluntary muscle control occurs as impulses from the motor cortex of the brain, travelling via the descending motor pathways, receive input from the cerebellum and basal ganglia before crossing over (decussation) at the medullary pyramids to travel down the spinal cord. The pyramidal tracts from the cortex exert voluntary control of motor function, while the extrapyramidal tracts from the other areas of the central nervous system have an inhibitory influence and control involuntary movement1,2,4.
SPINAL CORD
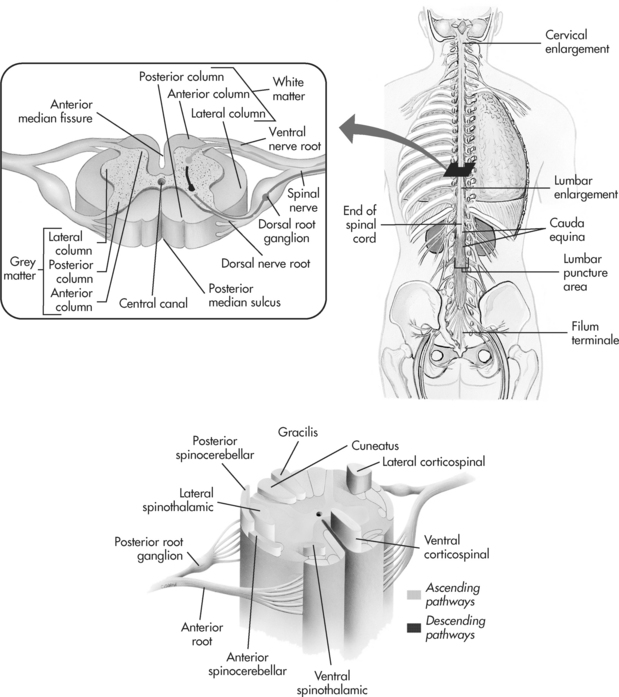
The spinal cord is continuous with the medulla oblongata to the level of the twelfth lumbar vertebra and consists of an inner mass of grey matter divided into horns containing the neuronal cell bodies which relay impulses related to reflex or voluntary motor activity. The surrounding white matter consists of myelinated nerve fibres grouped into columns called tracts that carry sensory or ascending impulses to the brain and descending or motor impulses to the nerve cells to refine voluntary muscle action. These messages travel from the spinal cord to the body via the 31 pairs of spinal nerves that are part of the peripheral nervous system (see Figure 8.4).
PERIPHERAL NERVOUS SYSTEM
The peripheral nervous system consists of 31 pairs of spinal nerves and their motor and sensory branches plus the 12 pairs of cranial nerves. The system has two divisions, the somatic and autonomic. The somatic division is involved with sensation and movement, while the autonomic nervous system (ANS) helps regulate the internal environment of the body through its effect on involuntary tissues in the lungs, and endocrine, cardiovascular and gastrointestinal systems. The autonomic nervous system includes the nerve cells and fibres that act upon these particular organs. It is subdivided into the sympathetic and the parasympathetic nervous system, each having antagonistic or opposing influences that help maintain homeostasis. The neurotransmitter of the sympathetic nervous system is noradrenaline and of the parasympathetic nervous system, acetylcholine. Control of the ANS is complex, involving the hypothalamus and brainstem structures4.
The 31 pairs of spinal nerves connect to the spinal cord by two roots. Sensory information is transmitted from receptors in the body to the dorsal nerve root of the nerve cell, and motor information is transmitted to the muscles via the ventral nerve root. Each spinal nerve separates into branches that supply a specific area, with a degree of overlap between the areas of the body each nerve supplies.
CEREBRAL BLOOD SUPPLY
The brain requires a blood flow of approximately 750mL/min for it to be able to function fully. The arteries and their branches in the brain receive blood supply from the right and left internal carotid arteries. These enter the skull anteriorly on each side through the base, and then branch to form the anterior and middle cerebral arteries that feed the anterior and middle part of the cerebral hemispheres. The posterior part of the cerebral hemispheres, including the occipital lobes, brain stem and cerebellum, receive their supply from the two vertebral arteries that enter at the foramen magnum to form the basilar artery, then this divides to form the two posterior cerebral arteries. The anterior and posterior communicating arteries join these two circulations to form a vascular ring called the circle of Willis. This potentially allows collateral circulation to develop if occlusion of a cerebral blood vessel occurs. Autoregulation in cerebral arterioles allows precise distribution of regional blood flow in the different areas of the brain3.
THE BLOOD BRAIN BARRIER
The blood brain barrier (BBB) maintains constancy in the intracerebral environment through controlling the passage of substances from the vascular system by a series of passive and selective active transport mechanisms. Substances such as oxygen, glucose, and essential nutrients enter the brain easily, while other substances are prevented from entering. This means that drugs require certain properties to allow them to cross the BBB if they are to act on the nervous system. For example, nicotine and alcohol enter easily while levodopa, the precursor of dopamine, is used to treat Parkinson’s disease because it can reach the brain from the vascular system, while dopamine is unable to enter. The protective effect of the BBB is diminished in the presence of cerebral disease, inflammation or trauma and certain organisms appear to be able to breakdown the barrier and cross into the nervous system4,5.
SUMMARY OF KEY CONCEPTS
GUILLAIN-BARRÉ SYNDROME
WHAT DOES IT MEAN?
Guillain-Barré syndrome (GBS) was named after two French doctors who first described a number of cases of acute ascending muscular weakness6. It is an acute immune-mediated disorder associated with inflammation and demyelination of the cranial and spinal nerves, characterised by a rapid progressive ascending muscle weakness resulting in varying degrees of motor dysfunction4,6,7. The syndrome occurs in children and adults, and is the most common cause of acute neuromuscular paralysis in developed countries8,9. There appear to be two major peaks of occurrence, in young adults and in persons aged over 55 years10,11. Although it can be fatal, prompt diagnosis and good management of symptoms leads to recovery of 80–90% of patients, with few or no remaining disabilities7,8.
WHAT IS THE PATHOPHYSIOLOGY?
GBS is believed to be caused by a cell-mediated autoimmune attack on the myelin surrounding the cranial and spinal nerves in response to a virus. Of patients with GBS, 50% have reported a viral-like illness 10–14 days prior to onset of symptoms7,12. Haemophilus influenzae, Mycoplasma pneumonia, Epstein-Barr virus, HIV and Camyplobacter jejuni infection have all been linked to GBS, as have patients who are immunocompromised, such as those who have AIDS or are post-organ transplant12–14.
In GBS, it is believed that an immunologic reaction results in inflammation and oedema of the myelin sheath of the nerve axon and the anterior and posterior nerve roots of the spinal nerve. In response, lymphocytes and macrophages move in to the area, which results in patchy loss of myelin (demyelination) from the affected peripheral nerves and wider spaces between the nodes of Ranvier. The more highly myelinated motor, and some cranial, nerves are affected more than those responsible for cutaneous pain, touch and temperature. In some cases, there may be injury to the axon itself4,6,11.
Demyelination causes impaired transmission of electrical impulses along the affected nerves due to the inability of the action potential to travel down the neuron and reach the target organ (loss of saltatory conduction). The result is a decrease in or loss of function in the organs the affected nerves innervate. The degree of dysfunction the patient experiences depends upon the degree of demyelination in the affected nerves. In motor nerves, muscle weakness and a flaccid paralysis (areflexia) can occur. Similarly, disruption of the posterior sensory nerve roots results in paresthesia, numbness, tingling and burning sensations, while the consequence of disruption to the autonomic nervous system may be a labile blood pressure and cardiac arrythmias4,6,11.
There is a range of variants of GBS based on the degree of peripheral nerve involvement or the site of inflammation, but ascending GBS or acute motor-sensory axonal neuropathy is the most common type, with symmetrical weakness and numbness starting in the legs and progressing to the trunk, arms and cranial nerves. Respiratory function is affected in 50% of cases8. Other variants are descending GBS, pure motor GBS or acute motor axonal neuropathy in which there is no sensory loss; and Miller-Fisher, a rare type, more common in China, Japan, Africa, and South America, which results in ophthalmoplegia, areflexia and ataxia8.
WHAT ARE THE CLINICAL MANIFESTATIONS?
The presenting signs and symptoms depend upon the degree of peripheral nerve involvement and the severity of the demyelination. Characteristically, the person will present with a progressive symmetrical ascending weakness of skeletal muscles. The weakness usually starts in the legs and may then affect the trunk, arms, and cranial nerves, with reduction in or loss of tendon reflexes8–14. In severe cases, respiratory failure may occur if the nerves supplying the intercostal muscles and diaphragm or the 10th cranial (vagus) nerve are affected. If the cranial nerves are involved, clinical signs will depend upon the nerve affected. For example, if the facial nerve is affected, there will be difficulty in smiling, frowning, or drinking with a straw. Dysphagia and paralysis of the larynx can occur due to involvement of the 9th, 10th, 11th and 12th cranial nerves8.
If there is sensory involvement, then paresthesia, sensations of tingling, altered sensation to touch and numbness in the extremities, reduced proprioception and sensory loss in a ‘stocking and glove’ distribution may be present. Pain and muscle stiffness in the limbs and back are also reported in about 25% of patients, and may become severe8.
Rapid shallow breathing with the use of the accessory muscles of respiration (abdominal, intercostal muscles, shoulder girdle) is present as the intercostal muscles and diaphragm become weaker. If vital capacity falls below 15mL/kg, there is a risk of respiratory failure and mechanical ventilation is indicated as alveolar hypoventilation and respiratory acidosis develop8,15. The most serious complication, respiratory failure requiring admission to ICU and mechanical ventilation, occurs in approximately 20–35% of cases16.
Autonomic dysfunction results in hypotension, hypertension or a labile blood pressure, and tachycardia, bradycardia or cardiac arrhythmias. Paralytic ileus, urinary retention, and inappropriate antidiuretic hormone secretion can also occur8,9.
GBS has three phases: acute, plateau and recovery. Symptoms usually occur 10–14 days following a viral infection and reach their peak in the third or fourth week. However in some cases, respiratory failure occurs within 48 hours of the onset of symptoms10,15. The acute phase lasts 1–3 weeks and begins when the first obvious symptoms develop, ending when no further symptoms appear or deterioration has ceased. The plateau phase, during which no change or improvement occurs, lasts from days to two weeks, while recovery time when remyelination and regrowth of axons occur can last from four months to three years. Approximately 80% of people recover from most effects by one year, although a 3% relapse rate has been reported after complete or near recovery9,15.
WHAT SHOULD YOU BE LOOKING AT IN LABORATORY AND OTHER TESTS?
Lumbar puncture and examination of the CSF will demonstrate an increase in protein over several days, peaking 4–6 weeks after onset of the disease, normal white cell count and some increase in CSF pressure. Blood tests may show an elevated erythrocyte sedimentation rate (ESR) and raised levels of immunoglobulin can be measured in the serum. Electromyography (EMG) and nerve conduction studies will demonstrate a slowing of the velocity of nerve conduction to voluntary muscle as demyelination occurs8,13.
WHAT IS THE TREATMENT?
Immunomodulation therapy has been found to prevent further progression of the disease and reduce recovery time if instigated within the first two weeks of the disease9. Indications for immunomodulation therapy are rapid progression of the disease, bulbar involvement, respiratory dysfunction, and/or loss of mobility. Intravenous immunoglobulin (IVIg) is the most commonly used treatment, as it is less invasive than plasmapheresis and is usually given over 2–5 days for a total dose based on body weight8. Plasmapheresis aims to remove circulating antimyelin antibodies, and a daily exchange of plasma is performed over 4–5 days17. However, it has been reported as less useful for the treatment of children, a proportion of who may have poorer long-term outcomes18–20.
APPLIED PHARMACOLOGY
Pharmacological management is based on immunomodulation therapy, prevention of complications and treatment of signs and symptoms. Anticoagulant therapy (heparin)21 is administered to prevent deep venous thrombosis and pulmonary embolus, both complications related to reduced venous return due to the flaccid paralysis and loss of normal muscle pump activity. Analgesia will be required if muscle pain is present and may include narcotics. Autonomic system manifestations, such as cardiac arrhythmias, bradycardia, and hypotension, may require appropriate medication to treat these symptoms. Neuromuscular blocking agents are to be avoided due to the risk of prolonging muscle paralysis.
NURSING IMPLICATIONS
Monitoring to detect signs of ascending paralysis and sensory loss are paramount. In particular, observing for poor airway clearance, respiratory failure, poor swallowing, and autonomic complications is essential to provide early treatment. Further, in determining the progress of the disease and preventing complications, assessment of motor and sensory function, pain, respiratory patterns, vital capacity, swallowing, cardiac rate and rhythm, blood pressure and urinary output are all important. Nursing interventions to prevent secondary complications, such as superimposed respiratory and urinary infection, limb contractures, bowel and bladder dysfunction, pressure ulcers, muscle atrophy, thrombo-embolic episodes and inadequate nutrition, are essential if the patient is to recover with minimal side effects. Psychological support, communication and education for both patients and their families are important to reduce anxiety and fear of the possible outcome.
MULTIPLE SCLEROSIS
WHAT DOES IT MEAN?
Multiple sclerosis (MS) is unpredictable inflammation, demyelination and scarring of the myelin in the brain, spinal cord and cranial nerves, resulting in widespread neurologic dysfunction. It is characterised by remissions and exacerbations of varying severity, with some patients having a rapidly progressive disease; however the majority (70%) have prolonged remissions22.
The incidence of disease is more common in people of northern European descent, in women born in the northern hemisphere from urban and high socioeconomic backgrounds, and there is often a family history8. Multiple sclerosis is a major cause of disability in people between 18–40 years, with average age at onset being 30 years8.
There is no one identified cause of MS and environmental agents, a slow acting virus, autoimmune response, allergic response, anoxia, toxins, and nutritional, traumatic and genetic factors have all been considered. The risk of developing the disease decreases in people who migrate from a high-risk area before adolescence, leading to a hypothesis that an environmental agent may trigger an autoimmune response in genetically susceptible people. An increase in humoral and cell-mediated T and B cells has been identified in the plaque22.
WHAT IS THE PATHOPHYSIOLOGY?
There is sporadic patchy demyelination of the white matter of the central nervous system in brain, spinal cord and cranial nerves, with a preference for the optic nerves, brain stem, cerebellum and spinal cord white matter secondary to inflammation4. Scarring from proliferation of the glial cells (gliosis) of the myelin sheath occurs in the affected areas with hard yellow plaques replacing the myelin. This scar tissue damages the axon fibre and causes disruption of the conduction of nerve impulses, usually in the brain, spinal cord or optic nerves. Nerve impulses may be slowed, blocked or be abnormal or ectopic, and this disruption accounts for the variation in symptoms with which patients present8.
Remission, with an improvement in symptoms, is thought to occur when the inflammation ceases or the lesions heal, with impulse transmission returning as remyelination by the oligodendrocytes occurs. Residual disability may be due to only part remyelination of lesions occurring. Symptoms become irreversible if further lesions develop, with permanent myelin loss and axonal loss if the disease progresses6,23.
Classification of multiple sclerosis is based upon the timing of exacerbations and disease progression, with remissions varying from months to years and relapses lasting days to months23,24 (see Table 8.1).
TABLE 8.1 TYPES AND CHARACTERISTICS OF VARIANTS OF MULTIPLE SCLEROSIS
| Type | Characteristics |
|---|---|
| Relapsing-remitting MS (RRMS) Most common | Recurrent episodes of neurological dysfunction with full, incomplete or no recovery. No progression of symptoms between attacks. 80% of cases are stable between attacks |
| Secondary progressive MS (SPMS) | Begins as RRMS but the number of attacks reduces and is replaced by a steady deterioration unrelated to attacks after twenty years or more |
| Primary progressive MS (PPMS) 10–15% | Older age onset. Characterised by a progressive course from the beginning signs and symptoms with no acute attack |
| Progressive relapsing MS (PRMS) 5% | Progressive deterioration from first symptoms, with an occasional acute attack |
WHAT ARE THE CLINICAL MANIFESTATIONS?
Clinical manifestations vary depending on the extent and location of demyelination, and therefore the varying presentations and unpredictable progress of the disease can make diagnosis difficult. The most common initial manifestations are visual and sensory alterations. Blurred vision and pain in the eye (optic neuritis), diplopia (double vision) and/or sensory disturbances, such as paresthesia, numbness and tingling, loss of balance, decreased ability to distinguish temperature and proprioception, are most commonly reported24.
Other signs and symptoms include emotional lability due to involvement of white matter in the frontal lobes. Gait ataxia, problems with balance and coordination, tremor, slurred speech and nystagmus occurs due to involvement of the cerebellum. Brain stem lesions affect swallowing and speech, while muscle weakness and spasticity are related to spinal cord tract involvement, as are bladder, bowel and sexual dysfunction24.
Approximately 65% of patients will have some form of cognitive impairment affecting functions such as memory, attention, information processing, executive function and visual spatial skills25. These are independent of disease progression and are often undiagnosed or mistaken for depression.
WHAT SHOULD YOU BE LOOKING AT IN LABORATORY AND OTHER TESTS?
There is no single test that diagnoses MS and initially, testing may not demonstrate any specific findings consistent with the disease. A negative test result does not rule out the diagnosis of multiple sclerosis, as it is based on the total picture rather than a single test23. Diagnosis is based on past and present patient history, full neurological examination including cranial nerves, motor, sensory and cerebellar function, and mental status. Magnetic resonance imaging (MRI) is the most sensitive test for multiple sclerosis, although CT scanning may show lesions in the white matter. MRI shows the size, number and location of lesions in the white matter brain or spinal cord in about 95% of cases. Evoked potential testing demonstrates slowed absent or abnormal conduction pathways of visual, auditory, sensory or motor nerve impulses in 85% of people23.
Components of the breakdown of myelin are released into the CSF as demyelination occurs and these components can be measured by three tests. The CSF IgG (immunoglobulin G) Index test detects IgG synthesis if the ratio of IgG: albumen is greater than the serum ratio. The normal IgG index is 0.77 or less, and the normal ratios of IgG: albumin in CSF is 0.15–0.38 and 0.15–0.41 in serum. The oligoclonal band test is used to examine if synthesis of immunoglobulin G (IgG) has occurred and is positive in more than 90% of patients with multiple sclerosis8. The third test, the CSF myelin basic protein test, is positive if elevated above 4ng/mL. However, as elevated levels occur in other demyelinating disorders, CNS trauma or infection, it cannot be used as the only test26.
Diagnosis is based on age, signs and symptoms of disease of the brain or spinal cord following neurological examination, evidence of 2 or more lesions on an MRI scan and a history of two or more episodes lasting at least 24 hours and occurring at least one month apart, or progressive development of signs and symptoms over at least 6 months with no other cause or explanation8,26.
WHAT IS THE TREATMENT?
There is no standard treatment to prevent or cure multiple sclerosis. The aims of treatment are to shorten the length of an attack, decrease demyelination, enhance recovery from an acute attack, and reduce the rate of relapse, as well as to slow disability and development of new lesions. This is achieved through pharmacological means and a multidisciplinary approach. The pharmacological approach includes the use of drugs that modify the immune processes (immunomodulators). Corticosteroids are used to reduce the severity and length of an acute attack or relapse by reducing oedema, and relieving symptoms. Interferons reduce the rate of relapse in relapsing-remitting MS (RRMS) and secondary progressive MS (SPMS) versions of the disease. Other drugs may be prescribed if the patient has a poor response or develops adverse reactions including chemotherapeutic agents, and humoral therapy through IVIG and plasmapheresis27–29.
Other management is focussed on managing the symptoms by supporting altered body functions, maintaining muscle tone and strength, improving function and preventing complications such as falls, constipation, urinary tract infection, joint contractures, pressure sores, and pneumonia through a multidisciplinary approach. Reduction of stress and prevention of fatigue are important components of management30,31. Physiotherapy for muscle strengthening and balancing exercises, spasticity reduction and development of a daily exercise routine plays a major role in management. Pain management may be required. Procedures to reduce severe spasticity include botulinum toxin injection, implantation of an intrathecal pump to administer the antispasmodic drug baclofen, or in extreme cases, surgical interventions such as adductor tendonotomy and dorsal rhizotomy. Deep brain stimulation has been used in cases where a disabling tremor is present26–27.
APPLIED PHARMACOLOGY
Corticosteroids are used to reduce the severity and length of an acute attack by reducing oedema, relieving symptoms, and shortening the length of an acute attack or relapse. A short course of intravenous (IV) methylprednisone (3–7 days) and an oral prednisone taper is the most commonly used approach.
A daily subcutaneous (SC) injection of glatiramer acetate (Caponex) is used for initial treatment of relapsing-remitting multiple sclerosis. Interferon–beta 1a (Avonex) via intramuscular injection (IMI) weekly, or interferon–beta 1b (Betaferon) SC every second day, is thought to increase protein synthesis, and decrease the frequency of acute attacks28. However, approximately 24–35% of patients will develop neutralising antibodies to these medications, therefore a decision about when to commence this treatment and close monitoring of its effect needs to be given serious consideration28,29.
Depending upon the signs and symptoms, a number of other medications may be prescribed. These include: antispasmodics for muscle spasticity (baclofen, diazepam or dantrolene sodium); anticholinergics for detrusor muscle spasm or urinary frequency and urgency (propantheline bromide); stool softeners, stimulants and bulking agents for bowel dysfunction; anticonvulsants (carbamazepine, gabapentin) for neuropathic pain; antidepressants; beta blockers or central nervous system depressants for tremor (propranolol, clonazepam); and amantadine which has been found to reduce fatigue27–31.



