21 Multiple Organ Dysfunction Syndrome
After reading this chapter, you should be able to:
• define the common terminology related to multiple organ dysfunction syndrome
• describe the related pathophysiology of multiple organ dysfunction syndrome
• identify the clinical manifestations of multiple organ dysfunction syndrome
• identify patients at risk of developing multiple organ dysfunction, including predictors of mortality
• initiate appropriate monitoring, care planning and evaluation strategies for the patient with multiple organ dysfunction in relation to the current evidence base
• discuss treatment strategies that promote homeostasis in the patient with multiple organ dysfunction syndrome
Introduction
The term multiple organ dysfunction syndrome (MODS) was established by an expert consensus conference in 1992 to describe a continuum of physiologic derangements and subsequent dynamic alterations in organ function that may occur during a critical illness.1,2 Previous terminologies in the literature were confusing. For example, multiple organ failure (MOF) was a term commonly used, but somewhat misleading as normal physiologic function can, in most cases, be restored in survivors of a critical illness who have temporary organ dysfunction.3,4 Although the syndrome affects many organs, it also affects physiological systems such as the haematological, immune and endocrine systems. MODS therefore more accurately describes altered organ function in a critically ill patient who requires medical and nursing interventions to achieve homeostasis.4
MODS is associated with widespread endothelial and parenchymal cell injury because of hypoxic hypoxia, direct cytotoxicity, apoptosis, immunosuppression and coagulopathy.4 Four clinical stages describe a patient with developing MODS:5
1. increasing volume requirements and mild respiratory alkalosis, accompanied by oliguria, hyperglycaemia and increased insulin requirements
2. tachypnoea, hypocapnia and hypoxaemia, with moderate liver dysfunction and possible haematological abnormalities
3. developing shock with azotaemia, acid–base disturbances and significant coagulation abnormalities
4. vasopressor dependence with oliguria or anuria, ischaemic colitis and lactic acidosis.
Cellular damage in various organs in patients who develop MODS begins with the onset of local injury that is then compounded by activation of the innate immune system. This includes a combination of pattern recognition, receptor activation and release of mediators at the microcellular level, leading to episodes of hypotension or hypoxaemia and secondary infections.4,5 The primary therapeutic goal for nursing and medical staff is prompt, definitive control of the source of infection or pro-inflammation6 and early recognition of preexisting factors that may lead to subsequent organ damage away from the initial site of injury. This preemptive therapy is instituted to maintain adequate tissue perfusion and prevent the onset of MODS. Recognition and response to early signs of clinical deterioration are therefore important to minimise further organ dysfunction.
This chapter initially describes the pathophysiology of inflammatory and infective conditions that may lead to multiple organ dysfunction. System responses and specific organ dysfunction are discussed, expanding on dialogue in previous chapters, particularly Chapters 19 and 20. Assessment of the severity of MODS and nursing considerations in the treatment of the MODS patient is presented.
Pathophysiology
The syndrome of multiple organ dysfunction is most closely related to an outcome of sepsis, which was described in Chapter 20. MODS is a state characterised by aberrant cellular responses involving multiple organ systems and sequential processes. The pathogenesis of MODS is complex, simultaneously involving every cell type, neuro-hormonal axis and organ system.7
In brief, hypoxic hypoxia results from altered metabolic regulation of tissue oxygen delivery which contributes to further organ dysfunction. Microcirculatory injury as a result of lytic enzymes, and vasoactive substances (nitric oxide, endothelial growth factor), is compounded by the inability of erythrocytes to navigate the septic microcirculation. Mitochondrial electron transport is affected by endotoxins in sepsis, nitric oxide and TNF-alpha, leading to disordered energy metabolism (see Figure 21.1). This causes cytopathic or histotoxic anoxia (the inability to use oxygen, even when available).8 This context of impaired oxygen utilisation rather than delivery7,8 results from diminished mitochondrial production of cellular energy (ATP), despite normal or even supranormal intra-cellular PO2 levels.9 Cytopathic hypoxia appears resistant to resuscitation measures, and this may ultimately worsen already-existing organ dysfunction. During sepsis or ischaemia, mitochondria respond by facilitating cell death rather than the restoration of homeostasis.7
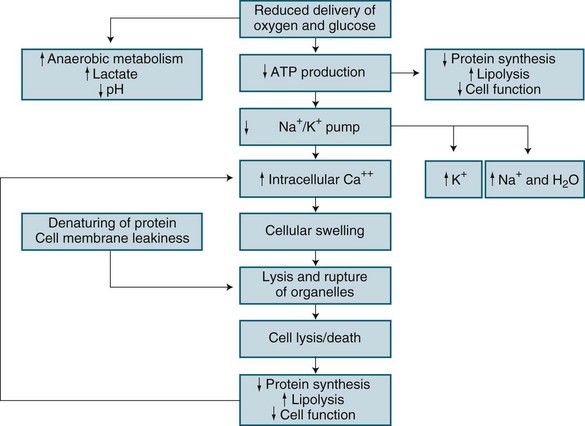
Apoptosis is normal physiological programmed cell death and is the main mechanism to eliminate dysfunctional cells.10 Apoptosis involves chromatin condensation, membrane blebbing, cell shrinkage and subsequent breakdown of cellular components into apoptic bodies. This normally orderly process is deranged in critical illness, leading to tissue or organ bed injury and MODS. Proinflammatory cytokines released in sepsis may delay apoptosis in activated macrophages and neutrophils, but in other tissues, such as gut endothelium, accelerated apoptosis occurs.8
In contrast, necrosis is a form of cell death characterised by cellular swelling and loss of membrane integrity as a result of hypoxia or trauma. Necrosis has been termed ‘cellular energy crisis’,10 and is unregulated resulting in loss of membrane sodium/potassium/ATP-ase pumps. This loss leads to cell swelling, rupture and spillage of intracellular contents into surrounding regions creating collateral damage.10 Necrosis therefore can involve significant amounts of tissue and organ bed damage. Apoptosis differs from necrosis in that it does not seem to involve the recruitment of inflammatory cells or mediators to complete its task. Activation of an enzyme cascade systematically cleaves proteins, including the cell’s nuclear DNA, with the end-result being death of the cell. This requires energy from mitrochondria and if not available necrosis of the cell occurs. Apoptosis and necrosis are processes that if is therefore important to understand in relation to future MODS research.
Increased concentrations of cell-free plasma DNA are present in various clinical conditions such as stroke, myocardial infarction and trauma, a likely result of accelerated cell death. Maximum plasma DNA concentrations correlated significantly with APACHE II scores and maximum SOFA scores (described later in this chapter), with cell-free plasma DNA concentrations higher in hospital non-survivors than in survivors. Using regression analysis, maximum plasma DNA was an independent predictor of hospital mortality.11
Other cellular organelles may also exhibit pathological reactions in MODS. In ischaemia/reperfusion, endoplasmic reticulum loses its ability to process proteins which induces the expression of heat shock proteins,7 affecting transcription of proteins necessary for organ specific functions. For example, liver cell metabolism, renal cell function or cardiac cell contractility may be affected.7 This has led to the controversial concept of a mode of hibernation of cells at the expense of survival of the whole organism.7
Cellular communication is also altered in MODS. Cells normally communicate through highly interactive bidirectional networks. The endothelium acts as a communication interface between cells, organs and systems and is involved in orchestration of systemic responses, including haemodynamic regulation, inflammation and coagulation; oxygen and nutrient delivery; oxidative stress and sensing of psychological stress and neuroendocrine alterations.7 In critical illness, endothelia release molecules that trigger the immune and neuroendocrine systems to produce a generalised inflammatory response.7 The combination of the pathophysiological processes involved with the development of MODS, compensatory mechanisms and the effect on target organs and systems is now discussed.
Systemic Response
After an overwhelming incident such as trauma, sepsis or non-infectious inflammation, a complex range of interrelated reactions occurs that result in a cascade of responses. The complex host-response generated involves the inflammatory immune systems, hormonal activation and metabolic derangements, resulting in multiple organ system involvement.12,13 These host-responses are initially adaptive to maintain nutrient perfusion to the tissues, however eventually organ systems become dysfunctional and fail, and the body is no longer able to maintain homeostasis16 (see Figure 21.2).
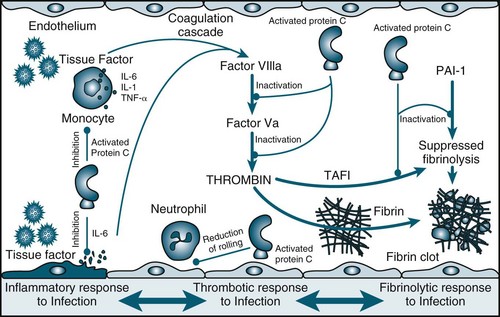
Initially, proinflammatory mediators are released locally to fight foreign antigens and promote wound healing. Antiinflammatory mediators are also released to downregulate the initial response to the insult.14 If the local defence system is overwhelmed, inflammatory mediators appear in the systemic circulation and recruit additional leucocytes to the area of damage. A whole-body stress response ensues, further compounding the situation. If proinflammatory mediators and antiinflammatory response is imbalanced, the patient may develop systemic inflammatory response syndrome (SIRS) and subsequent immunological dissonance15 of organ dysfunction.2,15,16
Intracellular transcription factors, in particular nuclear factor kappa B (NFκB), are important in innate and adaptive immunity,17,18 as they regulate the transcription of genes involved in the inflammatory and acute stress response, leading to expression of TNFα, interleukins and tissue factor.18,19 NFκB therefore plays an important role in response pathways in critical states including hypoxia, ischaemia, haemorrhage, sepsis, shock and MODS.18,20,21
The inflammatory cascade activates a number of prostaglandins and leucotrienes that also have pro- and antiinflammatory effects. Thromboxane A2 plays a role in the acute phase, in part due to stimulation of platelet aggregation, leading to microvascular thrombosis and tissue injury;15 it may also play a role in pulmonary bronchoconstriction and myocardial depression.
The specific pathophysiological concepts of inflammation, oedema and infection are discussed below.
Inflammation
Inflammation is part of innate immunity, a generic response to injury, and is normally an excellent mechanism to localise injury and promote healing.22,23 The basis of this immune response is recognition and an immediate response to an invading pathogen without necessarily having previous exposure to the pathogen.24 Neutrophils, macrophages, natural killer cells, dendrites, coagulation and complement are the principal active components of the innate host response.23
The classic signs of inflammation are:
Nitric oxide and prostaglandins (e.g. prostacyclin), are the primary mediators of vasodilation and inflammation at the injury site.23 Injured endothelium produces molecules that attract leucocytes and facilitate movement to the tissues. White blood cells accumulate by margination (adhesion to endothelium during the early stages of inflammation) and neutrophils accumulate at the injury site, where rolling and adherence to binding molecules on the endothelium occurs with eventual movement across the endothelium into the tissues.23 Different blood components therefore escape the intravascular space and occupy the interstitial space where they play the main role in successive phases of the inflammatory response. The endothelium therefore plays a bidirectional mediating role between blood flow and the interstitial space where inflammation mainly takes place.25 Macrophages, neutrophils and monocytes are responsible for phagocytosis and the production of toxic free radicals to kill invading pathogens.24 The complement system, a collection of 30 proteins circulating in the blood, is also activated, with plasma and membrane proteins acting as adjuncts to inflammatory and immune processes.26 When activated by inflammation and microbial invasion, these processes facilitate lysis (cellular destruction) and phagocytosis (ingestion) of foreign material.23,26
Dysfunction of organ systems often persists after the initial inflammatory response diminishes; this is largely unexplained, although dysoxia (abnormal tissue oxygen metabolism and utilisation) has been implicated.22,27 Hypoxia induces release of IL-6, the main cytokine that initiates the acute phase response. After reperfusion of ischaemic tissues, tissue and neutrophil activation forms reactive oxygen species (e.g. hydrogen peroxide) as a byproduct. These strong oxidants damage other molecules and cell structures that they form,23 resulting in water and sodium infiltrate and cellular oedema.
Oedema
Oedema occurs as a consequence of alterations to tissue endothelium, with increased microvascular permeability (‘capillary leak’). As noted earlier, many mediators, including circulating cytokines, oxygen free-radicals and activated neutrophils, alter the structure of endothelial cells, enabling larger molecules (proteins, water) to cross into the extravascular space.23,28 This response mechanism improves supply of nutrient-rich fluid to the site of injury, but if this becomes systemic, fluid shifts can lead to hypovolaemia, third-spacing (interstitial oedema) or affect other organs (e.g. acute lung injury, ALI).23
Infection and Immune Responses
Infection exists when there is one of the following: positive culture, serology,29 presence of polymorphonuclear leucocytes in a normally sterile body fluid except blood, and clinical focus of infection such as perforated viscus or pneumonia. In sepsis, the most common sites of infection are the lungs (34–54%), intra-abdominal organs (15–28%) and urinary tract (5–10%).30,31 The incidence of bloodstream infections is 30–40%,29 although one-third of cases with septic shock have negative blood cultures; one reason suggested for this is antibiotic administration prior to sample collection.32 The type of infecting organism has also changed over time, with Gram-positive bacteria predominant, accounting for at least one-third of pathogens in septic shock; Gram-negative, fungal, viruses and parasitic organisms are also involved.29 The increasing incidence of resistant organisms, partially as a result of the indiscriminate use of antibiotics, is an ongoing concern.
The immune response to infection has both non-specific and specific actions, with inflammation and coagulation responses intricately linked in sepsis pathophysiology.23,24,33,34 Tissue injury and the production of inflammatory mediators lead to:
• coagulation via the expression of tissue factor and factor VIIa complex (tissue factor pathway; the primary cascade for initiation of coagulation; previously termed the ‘extrinsic’ pathway)28,33–35
• coagulation amplification via factors Xa and Va, leading to massive thrombin formation and fibrin clots (common coagulation pathway).28,33
Note that blood cell injury or platelet contact with endothelial collagen initiates the contact activation pathway (previously termed the ‘intrinsic’ coagulation pathway).33
Procoagulation
Tissue factor is a procoagulant glycoprotein-signalling receptor,36 expressed when tissue is damaged or cytokines are released from macrophages or the endothelium (see Figure 21.3). Prothrombin is formed, leading to thrombin and fibrin generation from activated platelets. Resulting clots are stabilised by factor XIII and thrombin-activatable fibrinolysis inhibitor (TAFI).33,36 Fibrinolysis is a homeostatic process that dissolves clots via the plasminogen–tissue plasminogen activator (tPA)–plasmin pathway (involving antithrombin, activated protein C [APC] and tissue factor pathway inhibitor). APC:37
• reduces inflammation by decreasing TNF and NFκB production
• reduces thrombin production when activated via thrombin–thrombomodulin complexes (anticoagulant action)
• inhibits thrombin-activatable fibrinolysis inhibitor and plasminogen activator inhibitor-1 (profibrinolytic action).33,34
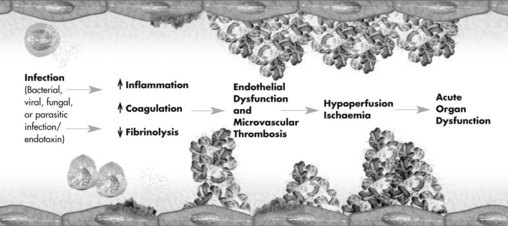
APC is consumed in severe sepsis, and thrombomodulin is unable to activate protein C,33,34,37 promoting a proinflammatory, prothrombotic state.34
Endocrine Response
Physiological changes are triggered as a normal response to a stressor. In a critically ill patient, however, chronic activation of the stress response, including the hypothalamic–pituitary–adrenal axis and the sympathetic–adrenal–medullary axis, results in ongoing production of glucocorticoid hormones and catecholamines.17 This response interferes with the regulation of cytokine-producing immune cells, leading to immune dysfunction. Other compensatory mechanisms are instigated in an attempt to maintain supply and perfusion to organs.15
These homeostatic mechanisms are activated through positive or negative feedback systems to counteract stress. When stress is extreme or prolonged, these normal homeostatic mechanisms may be insufficient and a patient may respond through a sequence of physiological changes called the stress response. The stress response occurs in three stages: the alarm reaction, the resistance reaction and exhaustion (see Table 21.1).38
TABLE 21.1 Actions of the stress response38
| Response stage | Neurohormonal response | Actions |
|---|---|---|
| Alarm reaction | Hypothalamus | |
| Noradrenaline/adrenaline | ||
| Resistance reaction | Anterior pituitary/corticotrophin-releasing factor | |
| Anterior pituitary/thyroid-stimulating hormone (TSH) | ||
| Human growth hormone (hGH) | ||
| ACTH | ||
| Kidney/renin release | ||
| Angiotensin II | ||
| Aldosterone | ||
| Exhaustion |
The alarm reaction (flight-or-fight response)38 is initiated when stress is detected, increasing the amount of glucose and oxygen available to the brain, skeletal muscle and heart. Two-thirds of total blood volume is also redistributed to support central circulation.38 A rise in glucose production and the breakdown of glycogen in skeletal muscle increases circulating glucose levels, providing an immediate energy source. The long-lasting second stage is a resistance reaction, involving hypothalamic, pituitary and adrenal hormone release.38 Response exhaustion occurs when these physiological changes can no longer maintain homeostasis.
Compensatory Mechanisms
Internal equilibrium (homeostasis) is maintained by the nervous and endocrine systems, and these work symbiotically with other compensatory mechanisms, such as endothelial cells, to maintain cellular perfusion. The nervous system responds rapidly to maintain homeostasis by sending impulses to organs to activate neurohormonal responses (see Chapters 16 and 20). Endothelins (ET-1, ET-2, ET-3) are potent vasoconstrictors produced by endothelial cells that regulate arterial pressure.20 The endocrine system works in a slow and sustained manner by secreting hormones, which travel via the blood to end-organs.
An initial acute-adaptive response is activated when an insult or stress occurs. For example, the body senses a disruption of blood flow through baroreceptor and chemoreceptor reflex actions: baroreceptors located in the carotid sinus detect changes in arterial pressure;13 chemoreceptors co-located with the baroreceptors detect O2, CO2 and H+ concentration. When alterations are sensed, the cardiovascular centre in the brain adjusts autonomic outflow accordingly.38 In a patient with decreased tissue perfusion, there is increased peripheral vasoconstriction, contractility and heart rate. Blood flow is shunted to the vital organs (brain, heart, lungs), and away from less vital areas (e.g. gastrointestinal and reproductive organs).39 Important hormonal regulators of blood flow are also activated from decreased blood flow to the kidneys, including adrenocorticotrophic hormone (ACTH), and the renin–angiotensin–aldosterone system (see Chapter 18). Adrenal medullary hormones, adrenaline and noradrenaline, vasopressin (antidiuretic hormone) and atrial natriuretic peptide also regulate blood flow to maintain adequate circulation and tissue oxygenation.13,38,39
Arterial pressure is a major determinant of tissue perfusion as it forces blood through the regional vasculature.20 Hypotension (systolic blood pressure <90 mmHg or mean arterial pressure [MAP] <70 mmHg) results from either low systemic vascular resistance or a low cardiac output.20 Glomerular filtration falls, leading to reduced urine output; low cerebral blood flow results in an altered level of consciousness; and other manifestations reflect low-flow states in other organ systems. To maintain oxygen supply, respirations and heart rate increase to meet organ oxygenation demands.40 Organ dysfunction ensues if balance is not sufficiently restabilised (see Table 21.2).
TABLE 21.2 Acute organ dysfunction46,98
| Organ system | Clinical parameters |
|---|---|
| Cardiovascular | Patient requires vasopressor support (systolic BP <90 mmHg) or MAP <70 mmHg for 1 hour despite fluid bolus |
| Respiratory | Patient requires mechanical ventilation: P/F ratio <250, PEEP >7.5 cmH2O |
| Renal | Low urine output <0.5 mL/kg/h; raised creatinine >50% from baseline or requiring acute dialysis |
| Haematological | Low platelet count (<1 000 000/mm3) or APTT/PTT > upper limit of normal |
| Metabolic | Low pH with increased lactate (pH <7.3 and plasma lactate > upper limit of normal) |
| Hepatic | Liver enzymes >2 × upper limit of normal |
| CNS | Altered level of consciousness/reduced Glasgow Coma Scale score |
| Gastrointestinal | Translocation of bacteria, possible elevated pancreatic enzymes and cholecystitis |
Organ Dysfunction
Organ dysfunction is a common clinical presentation in ICU. Patients with dysfunction in the respiratory, cardiovascular, hepatic or metabolic systems were 50% more likely to require ICU treatment and had a higher mortality than patients not requiring intensive care.41 Timely identification of organ dysfunction is therefore critical, as early intervention reduces damage and improves recovery in organ systems. As each organ fails, the average risk of death rises by 11–23%, with up to 75% of patients in sepsis clinical trials having at least two failing organs.42 The organ system that most commonly fails is the pulmonary system, followed by the cardiovascular, renal and haematological systems.43 Organ and systems dysfunction are a result of hypoperfusion, inflammation, cellular dysfunction and oedema. Dysfunction of the cardiovascular (Chapters 10 and 12), respiratory (Chapters 14 and 15), renal (Chapter 18), and hepatic and gastrointestinal systems (Chapter 19) have been previously addressed. This next section addresses the haematological, endocrine and metabolic systems. Neurological dysfunction is also common in the patient with MODS and complements previous discussions in Chapter 17.
Haematological Dysfunction
Systemic inflammatory response syndrome (SIRS) and disseminated intravascular coagulation (DIC) have pivotal and synergistic roles in the development of MODS.44 The coagulopathy present in MODS results from deficiencies of coagulation system proteins (e.g. protein C, antithrombin 3 and tissue factor inhibitors).8 Inflammatory mediators initiate direct injury to the vascular endothelium, releasing tissue factor, triggering the extrinsic coagulation cascade and accelerating thrombin production.8 Coagulation factors are activated as a result of endothelial damage with binding of factor XII to the subendothelial surface, activation of factors XI, XII, X, VIII, calcium and phospholipid.8 The final pathway is production of thrombin which converts soluble fibrinogen to fibrin. Fibrin and aggregated platelets form intravascular clots.
Inflammatory cytokines also initiate coagulation though activation of tissue factor (TF), a principal activator of coagulation. Endotoxins increase the activity of inhibitors of clot breakdown (fibrinolysis). Levels of protein C and endogenous activated protein C are decreased in sepsis; this inhibits coagulation cofactors Va and VIIa and acts as an antithrombotic in the microvasculature.8
Microvascular thrombosis that leads to MODS results from two major syndromes: thrombotic microangiopathy (TMA) and disseminated intravascular coagulation (DIC). TMA is characterised by formation of microvascular platelet aggregates and occasionally fibrin formation. Typically there is history of injury to the microvascular endothelium (e.g. thrombotic thrombocytopenic purpura, haemolytic uremic syndrome, haemolytic anaemia, elevated liver enzymes and low platelet syndromes of pregnancy or antiphospholipid antibody syndrome).44 TMA usually presents with normal coagulation profiles such as prothrombin times and partial thromboplastin time.44
Disseminated intravascular coagulation results from widespread activation of tissue factor-dependent coagulation, insufficient control of coagulation and plasminogen-mediated attenuation of fibrinolysis.44 This leads to formation of fibrin clots, consumption of platelets and coagulation proteins, occlusion of the microvasculature, and resultant reductions in cellular tissue oxygen delivery.44 DIC is most commonly a result of trauma or sepsis and is an exaggerated response to normal coagulation aimed at limiting infection, exsanguination and promoting wound healing.44
Thrombocytopenia (a platelet count of <80,000/mm3 or a decrease of ≥50% over the preceding three days) signifies haematological failure,45 with leucocytopenia/cytosis, markers of coagulation and DIC also present.46 Treatment is supportive and aimed at removing the triggering insults. Clinical biomarkers include a simultaneous rise in prothrombin time, APTT and thrombocytopenia.34 A patient may exhibit bleeding from puncture sites (e.g. invasive vascular access), mucous membranes including bowel, or upper gastrointestinal tract. Bruising or other subcutaneous petechiae may be evident. The skin should be protected from trauma.
Primary therapy is directed at the cause of the insult, with SIRS, ischaemia, uraemia, hepatotoxins and sources of infection, injury or necrosis managed concurrently. Aggressive resuscitation includes crystalloid or colloid administration, replacement of blood components and clotting factors using packed cells, platelets, cryoprecipitate and fresh frozen plasma. Endpoints for haemoglobin, platelets and coagulation levels have not been agreed upon and replacement is therefore individualised.47
The role of heparin or fractionated heparin is controversial in the presence of sepsis, particularly in those with overt thromboembolism or extensive fibrin deposition, such as in purpura fulminans or ischaemia in the extremities.47,48 Administration of APC in its role as inhibitor of the coagulation cascade is controversial. A Cochrane review of four studies involving 4911 participants (4434 adults and 477 paediatric patients) identified no reduction in risk of death (28-day mortality) in adult participants with severe sepsis, but was associated with a higher risk of bleeding. Effectiveness was not associated with the degree of severity of sepsis.49 Studies continue into this area of clinical practice.
Endocrine Dysfunctions
Numerous endocrine derangements are noted in critically ill patients, including abnormalities in thyroid, adrenocortical, pancreas, growth and sex hormones. A high thyrotropin (TSH) level is a significant independent predictor of non-survival in critically ill patients,50 while subclinical hypothyroidism has significant negative effects on cardiac function and haemodynamic instability.50,58
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


