Chapter 3 Molecular biology of HIV
implications for new therapies
HIV Entry
The 9-kilobase HIV RNA encodes nine genes, yielding 15 distinct proteins. Compared to other retroviruses, HIV is genetically complex. Investigations over the past several years yielded informative insights into the function of each of HIV’s individual gene products, many of which could form potential targets for new therapies (Fig. 3.1). To productively infect a cellular target, HIV must introduce its genetic material into the cytoplasm of this cell. HIV entry requires the initial binding or attachment of the HIV envelope protein to (Env) CD4 receptors on the surface of target cells. The rational development of inhibitors of HIV attachment has been propelled by structural studies unraveling the “lock and key” assembly of trimeric gp120 Env spikes present on virions with CD4 receptors residing on the surface of the target cells [1]. The insertion of a key phenylalanine residue in the outer portion of the CD4 protein into a recessed pocket in gp120 produces a very high-affinity interaction of these two proteins.
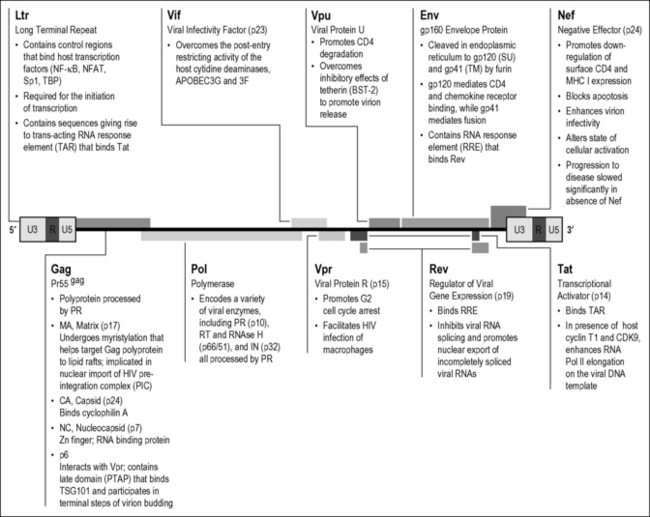
BMS 488043 is a promising second-generation attachment inhibitor that blocks the insertion of this CD4 residue into this pocket of gp120. Protein-based approaches to interrupting the gp120–CD4 interaction are also being developed. Numerous studies have identified a handful of monoclonal antibodies against HIV-1 that neutralize primary isolates of HIV-1 in vitro and prevent infection in non-human primate models [2–4]. Although these antibodies clearly prevent infection in animal models, very high doses of the antibodies are required for clinical effects in HIV-infected patients. Other protein-based therapies include PRO 542, a tetravalent fusion protein containing the D1 and D2 domains of human CD4 and the heavy- and light-chain constant regions of human IgG2 [5, 6]. Phase I and II clinical trials revealed modest decreases in viral load in patients with advanced disease [7]. Another candidate is ibalizumab (formerly TNX-355), a humanized anti-CD4 IgG4 monoclonal antibody that specifically reacts with a conformational epitope in the D2 domain of CD4 induced by gp120 binding. This antibody blocks subsequent Env engagement of the HIV chemokine co-receptors [8]. The antibody showed beneficial activity relative to optimized background therapy in patients and has advanced to phase II clinical trials [9].
The second phase of HIV entry involves the engagement of the HIV co-receptors. The initial binding of trimeric gp120 to CD4 induces a conformational change in the envelope that promotes binding of the virion to a specific subset of chemokine co-receptors. These receptors contain seven membrane-spanning domains and normally help hematopoietic cells to migrate down specific chemokine gradients to sites of inflammation. Although these receptors signal through G proteins [10], such signaling is not required for HIV infection. Twelve different chemokine receptors function as HIV co-receptors in cultured cells, but only two, CCR5 and CXCR4, are normally used in vivo [10]. CCR5 binds macrophage-tropic, non-syncytium-inducing (R5-tropic) viruses, which are associated with mucosal and intravenous transmission of HIV infection. CXCR4 binds T-cell-tropic, syncytium-inducing (X4-tropic) viruses, which generally emerge only during the later stages of disease [11]. Utilization of CXCR4 is associated with accelerated disease progression but only occurs in about half of patients infected with HIV.
A naturally occurring deletion of 32 base pairs in the CCR5 gene [12, 13] is present in approximately 13% of individuals of northern European descent. This mutation gives rise to a truncated form of the CCR5 receptor that never reaches the cell surface. Emphasizing the key role of CCR5 in horizontal transmission, individuals homozygous for this CCR5 Δ32mutation (1–2% of the Caucasian population) are almost completely resistant to HIV infection [12, 13]. This “experiment of nature” propelled pharmaceutical development of small molecules that prevent HIV interaction with CCR5. Within only 7 years after the discovery of HIV co-receptors, several small-molecule antagonists of CCR5 entered clinical use. Maraviroc (Selzentry) is a low-molecular-weight CCR5 antagonist used for treatment of HIV-infected persons harboring R5 viruses resistant to conventional drugs [14]. Treatment of therapy-naïve patients with Maraviroc is still being analyzed in clinical trials. TAK-220, another low-molecular-weight compound, targets a pocket between the transmembrane helicies of CCR5, thereby altering the conformation of the receptor, and has advanced to phase I clinical trials [15]. Protein-based CCR5 inhibitors include PRO 140, a humanized mouse antibody that recognizes an epitope at the CCR5 N-terminus, which has entered phase II clinical trials [16]. Interest has also focused on using modified versions of the chemokine RANTES, a natural ligand for CCR5, to block HIV infection. A key unanswered concern is whether blockade of CCR5 will promote an earlier switch to CXCR4 co-receptor utilization by HIV. As noted, such a switch could lead to more rapid clinical deterioration since many more CD4 T cells express CXCR4 than CCR5.
Both CD4 and chemokine co-receptors for HIV are found disproportionately within lipid signaling rafts located within the cell membrane [17]. These cholesterol- and sphingolipid-enriched microdomains likely provide a better environment for membrane fusion possibly because HIV virions bud from such lipid rafts and acquire similar lipids [18]. Removing cholesterol from virions, producer cells, or target cells greatly decreases the infectivity of HIV [19]. Studies are under way exploring whether cholesterol-depleting compounds might be efficacious as topically applied microbicides to inhibit HIV transmission at mucosal surfaces. The development of effective microbicides would form an exceptionally valuable approach in efforts to prevent HIV transmission. Approximately 50 microbicide candidates are in different stages of development in the microbicide pipeline. Thus far, mostly non-specific microbicides, such as surfactants and polyanions, have completed phase III clinical testing, and none has demonstrated clear statistical evidence of protection [20]. Tenofovir, a nucleotide analogue reverse transcriptase inhibitor, is the most advanced clinical candidate. A 1% vaginal gel formulation of tenofovir reduced HIV acquisition by nearly 40% overall in the recently completed CAPRISA phase II trial in South African women [21].
The third phase of HIV entry is virion fusion. The binding of surface gp120, CD4, and the chemokine co-receptors generates a sharp conformational change in gp41, the second HIV envelope protein [22]. Assembled as a trimer on the virion membrane, this coiled-coil protein springs open, projecting three peptide fusion domains that “harpoon” the lipid bilayer of the target cell. The gp41 trimers then fold back on themselves, forming a hairpin structure. The recently approved T20 fusion inhibitor, enfuvirtide, prevents the formation of the hairpin structure essential for successful fusion. The fusion reaction leads to intracytoplasmic insertion of the HIV viral core [22].
HIV virions also enter cells by endocytosis. However, this form of entry does not lead to productive viral infection likely because the internalized virions are inactivated within acidified endosomes. However, a special form of endocytosis associated with facilitated infection of CD4 T cells has been described in dendritic cells. These cells normally process and present antigens to immune cells, and many dendritic cells express a specialized attachment receptor termed DC-SIGN [23] or closely related C-type lectin receptors. DC-SIGN binds HIV gp120 with high affinity but does not trigger the conformational changes in env required for fusion. Rather, virions bound to DC-SIGN may be internalized into a vesicular compartment that does not lead to viral inactivation. These vesicles containing viable HIV virions are transported back to the cell surface after the dendritic cell has matured and migrated to regional lymph nodes, where it engages T cells [24]. Indeed, these virus-laden vesicles may selectively accumulate at the immunological synapse formed between dendritic cells and CD4 T cells. Thus, dendritic cells expressing DC-SIGN or related C-type lectin receptors may act as “Trojan horses,” facilitating the spread of HIV from mucosal surfaces to lymphatic organs in the absence of productive infection of the dendritic cell itself.
Recent studies describe an innate recognition pathway for HIV-1 in dendritic cells [25]. HIV-1 does not replicate in dendritic cells; however, this block can be overcome by the accessory protein Vpx from simian immunodeficiency virus (SIVmac). When this block is bypassed, HIV-1 replaces dendritic cell activation and the production of type I interferons. HIV-1-mediated activation of dendritic cells induced virus-specific IFN-γ-producing T cells and caused proliferation of CD4 T cells. Activating the HIV-1 sensor in dendritic cells could potentially improve the effectiveness of current vaccine candidates.
Early Cytoplasmic Events
HIV-1 contains an internal viral RNA–protein complex surrounded by a protein shell, termed the capsid core [26]. Upon viral fusion, the capsid core is released into the cytoplasm of the target cell. Although the ensuing stages are poorly understood, the capsid core is thought to undergo a controlled disassembly reaction. This process appears to involve phosphorylation of the matrix protein by a mitogen-activated protein (MAP) kinase [27] and additional actions of cyclophilin A [28] and the viral proteins Nef [29] and Vif [30]. Optimal stability of the capsid core appears to be critical for successful infection of the host cell. Mutations that alter the stability of the core, by rendering the particle either too stable or unstable, impair infection [31]. Thus, small molecules that perturb the structure and/or disassembly process may have therapeutic benefit. One recently identified candidate, PF-3450074 (PF74), appears to trigger premature uncoating of the capsid core, leading to an inability to successfully complete subsequent steps in the viral life cycle [32].
After the viral core is uncoated, the viral reverse transcription complex is liberated and begins the conversion of viral RNA into double-stranded DNA [33]. This complex includes the diploid viral RNA genome, tRNALys primer, reverse transcriptase, integrase, matrix, nucleocapsid, viral protein R (Vpr), and various host proteins. It docks with actin microfilaments [34]. This interaction, mediated by the phosphorylated matrix, is required for the commencement of efficient reverse transcription. A variety of nucleoside (AZT, ddI, ddC, and 3TC), nucleotide (tenofovir), and non-nucleoside (nevirapine) inhibitors of the HIV reverse transcriptase are widely used in the clinic. Indeed, targeting of the viral reverse transcriptase represents one of the most successful strategies for impairing HIV growth. Effective reverse transcription yields the HIV preintegration complex (PIC), composed of double-stranded viral cDNA, integrase, matrix, Vpr, reverse transcriptase, and the high mobility group DNA-binding protein HMGI(Y) [35]. The PIC may move toward the nucleus by sliding down microtubules [36, 37]. Adenovirus and herpes simplex virus 1 similarly dock to microtubules and use the microtubule-associated dynein molecular motor for cytoplasmic transport. This finding suggests that many viruses utilize these cytoskeletal structures for directional movement. Overexpression of FEZ1, a regulator of cytoskeletal transport, blocks both HIV-1 and murine leukemia virus infection at a step after reverse transcription [38]. Thus, overexpressed FEZ1 might disrupt normal virus trafficking.
At least two host proteins associate with the PIC and are important for its function. One is BAF-1 (barrier to autointegration factor-1), a small DNA-binding protein [39]. Removal of BAF-1 promotes the suicidal pathway of autointegration, which occurs when the 3’-ends of the reverse transcript attack sites within the viral DNA. The other host protein is LAP2α, a laminin-assocated component of the nuclear envelope, which promotes productive PIC integration [40].
Crossing the Nuclear Pore
Unlike most animal retroviruses, HIV can infect nondividing cells, such as terminally differentiated macrophages [41]. This requires an ability of the viral PIC to cross intact nuclear membranes. With a Stokes radius of approximately ~28 nm, resembling the size of a ribosome [42], the PIC is about twice as large as the maximal diameter of the central aqueous channel of the nuclear pore. It seems likely that the 3-μm contour length of viral DNA must undergo significant compaction, and the import process must involve considerable molecular gymnastics.
One of the more controversial areas of HIV research involves the identification of key viral proteins that mediate the nuclear import of the PIC. Integrase [43], matrix [44], and Vpr [45] have each been implicated (Fig. 3.2). Because plus-strand synthesis is discontinuous in reverse transcription, a triple helical DNA domain or “DNA flap” that may bind a host protein containing a nuclear targeting signal is produced [46]. Matrix encodes a canonical nuclear localization signal that is recognized by the importins-α and -β, which are key components in the classical nuclear import pathway. However, recent studies questioned the contributions of the nuclear import signal in integrase as well as the DNA flap to the nuclear uptake of the PIC [47]. The HIV Vpr gene product [48] contains at least three noncanonical nuclear targeting signals. Vpr may bypass the importin system altogether, perhaps mediating the direct docking of the PIC with one or more components of the nuclear pore complex. The multiple nuclear targeting signals within the PIC, which could involve yet-to-be-identified cellular factors, may also function in a cooperative manner or play larger roles individually in different target cells. For example, while Vpr is not needed for HIV infection of nondividing, resting T cells [49], it markedly enhances viral infection in nondividing macrophages [50]. The finding that both matrix [51] and Vpr [48] shuttle between the nucleus and cytoplasm likely ensures their availability for incorporation into new virions.
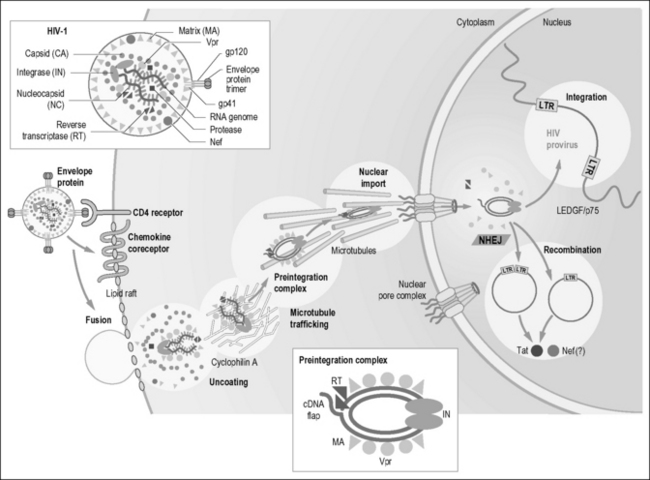
Host factors involved in nuclear import of the PIC also remain to be identified. One report implicated importin 7, one of a family of nuclear import receptors responsible for the uptake of ribosomal proteins and histones [52]. Another report suggested Nup98, a component of the nuclear pore complex, could be required for HIV-1 infection [53]. More recent work suggests small host RNAs, such as specific tRNAs, bind to the PIC and facilitate its entry into the nucleus [54]. However, the roles of these factors have yet to be conclusively demonstrated.
Integration
Once inside the nucleus, the viral PIC establishes a functional provirus (Fig. 3.2). Effective integration of the double-stranded viral DNA into the host chromosome is mediated by the HIV integrase, which binds the ends of the viral DNA [35]. The host proteins HMGI(Y) and barrier to autointegration (BAF) are required for efficient integration, although their precise functions remain unknown [55]. Integrase removes nucleotides from viral ends, producing a two-base recess and thereby correcting the ragged ends generated by the terminal transferase activity of reverse transcriptase [35]. It also catalyzes the subsequent joining reaction that establishes the HIV provirus within the chromosome. Recent studies indicate that HIV integration preferentially occurs in actively transcribed genes [56].
The search for integrase inhibitors has been slow. Only one integrase inhibitor, raltegravir (Isentress 1), is approved for clinical use [57]. Raltegravir, as well as similar compounds, selectively blocks DNA strand transfer activity and is therefore referred to as integrase strand transfer inhibitors (INSTIs). Resistance to raltegravir develops rapidly: a single mutation is sufficient to confer resistance [58].
One host factor required for HIV-1 integration is LEDGF/p75 (lens epithelium-derived growth factor), a nuclear transcriptional co-activator [59]. LEDGF/p75 binds tightly to HIV-1 integrase and appears to act as a chromatin-associated receptor for preintegration complexes [60, 61]. In cells devoid of endogenous LEDGF/p75 protein, both the levels of HIV-1 integration and their genomic distribution are significantly perturbed [62, 63].
Another host factor that seems to mediate the association of the preintegration complex with chromatin after nuclear entry is emerin, a component of the inner nuclear membrane [64]. The association of emerin with the PIC is mediated by BAF, and both proteins appear to cooperate to promote HIV integration into chromatin.
Not all PICs that enter the nucleus result in a functional provirus. The ends of the viral DNA may be joined to form a 2-LTR (long terminal repeat) circle or the viral genome may undergo homologous recombination yielding a single LTR circle. Finally, the viral DNA may autointegrate into itself, producing a rearranged circular structure. Although some circular forms may direct the synthesis of the transcriptional transactivator Tat and Nef, none produces infectious virus [65]. The nonhomologous end-joining system may form 2-LTR circles to protect the cell [66]. This system is responsible for rapid repair of double-strand breaks, which minimizes the number of free DNA ends within the cell, thereby preventing an apoptotic response. A single double-strand break detected within a cell is sufficient to induce G1 cell-cycle arrest. The ability of the free ends of the viral DNA to mimic such double-strand chromosomal breaks may contribute to the direct cytopathic effects observed with HIV.
Transcriptional Events
Integration can lead to latent or transcriptionally active forms of viral infection [67]. The chromosomal environment at the site of viral integration likely helps shape the provirus’s transcriptional activity [68]. For example, proviral integration into repressed heterochromatin might favor the generation of latent proviruses (Fig. 3.3). In addition, the transcriptional status of HIV-1 is tightly coupled to the activation state of the cell. In resting T cells, nucleosomes adjacent to the HIV promoter (e.g., Nuc 1) bear the characteristic marks of silent heterochromatin, such as lysine 9 trimethylated histone 3, heterochromatin protein 1, and low levels of histone acetylation [69]. Furthermore, the 5’ LTR of HIV can bind negative regulators of transcription, such as p50 homodimers (inactive form of nuclear factor κB, NF-κB) or the C-promoter binding factor 1 [70]. Both of these factors recruit histone deacetylases that act on Nuc1. Silencing is reinforced by methylation of two CpG islands and the subsequent binding of Methyl-CpG binding domain protein 2 (MDB2) [71, 72]. Finally, latency can be enhanced by post-transcriptional mechanisms, such as impaired HIV mRNA nuclear export and the expression of host micro-RNAs.
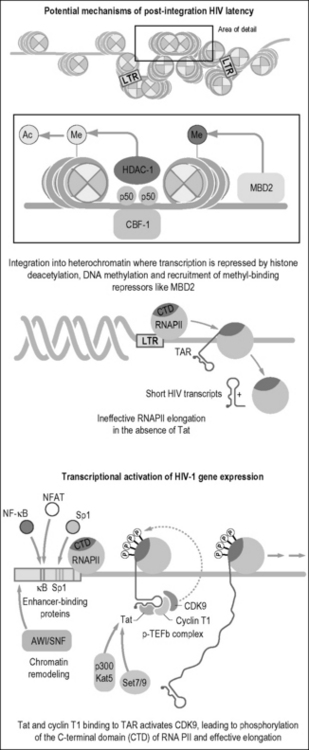
Because antiretroviral therapy can control, but not cure HIV infection, recent efforts have focused on the possibility that latent viruses can be reactivated and then purged by pharmacological manipulations. Three types of agents, all aimed at reactivating latent viruses, have been tested: T cell activators, inhibitors of histone-modifying enzymes, and inhibitors of DNA methylation [73]. Clinical trials have assessed T cell activators such as IL-2, IL-7, and antibodies specific to the T cell receptor CD3 subunit. Although these therapies transiently reduced the latent virus reservoir, patients generally experienced rapid viral rebound upon antiretroviral therapy cessation [74, 75]. In other studies, valproic acid, a weak inhibitor of histone deacetylase activity, showed some ability to decrease the pool of latently infected resting CD4 T cells; however, these effects have not been confirmed [76, 77]. In vivo studies have shown induction of transcriptionally repressed latent viruses using a combination of activators (e.g. the NF-κB inducer prostratin) in combination with inhibitors of histone deacetylation (e.g. valproic acid and suberoylanilide hydroxamic acid) and DNA methylation (e.g., 5-aza-2’-deoxycytidine) [71, 74, 78]. The combination of drugs aimed at purging latent reservoirs of virus may also activate endogenous retroelements. Thus, the benefits of reactivating latent proviruses must be weighed against the risk of retrotransposition-induced insertional mutagenesis.
In the host genome, the 5’ LTR functions like other eukaryotic transcriptional units. It contains downstream and upstream promoter elements, which include the initiator (Inr), TATA-box (T), and three Sp1 sites [79]. These regions help position the RNA polymerase II (RNAPII) at the site of initiation of transcription and assemble the preinitiation complexes. Transcription begins, but the polymerase fails to elongate efficiently along the viral genome (Fig. 3.3). In the process, short nonpolyadenylated transcripts are synthesized, which are stable and persist in cells due to the formation of an RNA stem loop called the transactivation response (TAR) element [80]. Slightly upstream of the promoter is the transcriptional enhancer, in which HIV-1 binds NF-κB, nuclear factor of activated T cells (NFAT), and Ets family members [81]. NF-κB and NFAT relocalize to the nucleus after cellular activation. NF-κB is liberated from its cytoplasmic inhibitor, IκB, by stimulus-coupled phosphorylation, polyubiquitylation, and proteasomal degradation of the inhibitor [82]. NFAT is dephosphorylated by calcineurin (a reaction inhibited by cyclosporin A) and, after its nuclear import, assembles with AP1 to form the fully active transcriptional complex [83]. NF-κB, which is composed of p50 and p65 (RelA) subunits, increases both the rates of initiation and elongation of viral transcription [84]. Since NF-κB is activated after several antigen-specific and cytokine-mediated events, it can stimulate transcription of silent proviruses primarily through the recruitment of histone acetyltransferases that remodel Nuc1
Tat is responsible for markedly increasing the rate of viral gene expression. With cyclin T1 (CycT1), it binds TAR and recruits the cellular cyclin-dependent kinase 9 (Cdk9) to the HIV LTR (Fig. 3.3) [85]. In the positive transcription elongation factor b (P-TEFb) complex, Cdk9 phosphorylates the C-terminal domain of RNAPII, which marks the transition from initiation to elongation of eukaryotic transcription [86]. Other targets of P-TEFb include negative transcription elongation factors (N-TEF), such as the DRB-sensitivity inducing (DSIF) and negative elongation (NELF) factors [86]. P-TEFb can be found in two distinct complexes, a small P-TEFb that contains only cdk9 and cyclin T, and a large P-TEFb that also contains 7SK small nuclear RNA and HEXIM1 (hexamethylene bisacetamide-induced protein 1).
In the absence of Tat, the HIV LTR functions as a very poor promoter because it so effectively binds these negative transcription factors in vivo. An arginine-rich motif (ARM) within Tat binds to the 5´ bulge region in TAR. A shorter ARM in cyclin T1, which is also called the Tat-TAR recognition motif (TRM), engages the central loop of TAR [85]. These regions form a high-affinity RNA-binding unit that is required for Tat transactivation. In the presence of the complex between Tat and P-TEFb, the RNAPII becomes a highly efficient elongating complex. Tat also recruits the SWI/SNF chromatin remodeling complex to the HIV promoter [87–89] to relieve the elongation block imposed by repressive nucleosomes.
Because murine CycT1 contains a cysteine at position 261, the complex between Tat and murine P-TEFb binds TAR weakly [90]. Thus, Tat transactivation is severely compromised in murine cells. Cdk9 also must be autophosphorylated on several serines and threonines near its C-terminus for productive interactions among Tat, P-TEFb, and TAR [91]. Additionally, basal levels of P-TEFb may be low in resting cells or only weakly active due to the interaction between P-TEFb and 7SK RNA [92].
Post-translational modifications of Tat, such as phosphorylation, methylation, and acetylation, modify its function and allow it to specifically interact with a wide array of cellular partners. For example, the lysine methyltransferase Set7/9 associates with the HIV promoter and monomethylates lysine 51, a highly conserved residue located in the RNA-binding domain of Tat [93]. Several histone-modifiying enzymes, such as p300/CBP, also associate with Tat. These enzymes acetylate Tat at lysines 50 and 51, promoting the disassociation of Tat from the TAR RNA and leading to the start of transcriptional elongation [94–96].
Expression of Viral Genes
Transcription of the viral genome results in more than a dozen different HIV-specific transcripts [97]. Some are processed cotranscriptionally and, in the absence of inhibitory RNA sequences (IRS), transported rapidly into the cytoplasm [98]. These multiply spliced transcripts encode Nef, Tat, and Rev, the “early” expressed genes of HIV. Other singly spliced or unspliced viral transcripts remain in the nucleus and are relatively stable. These viral transcripts encode the structural, enzymatic, and accessory proteins and represent viral genomic RNAs needed for the assembly of fully infectious virions.
Incomplete splicing likely results from suboptimal splice donor and acceptor sites in viral transcripts. In addition, the regulator of virion gene expression, Rev, may inhibit splicing by its interaction with alternate splicing factor/splicing factor 2 (ASF/SF2) [99] and its associated p32 protein [100].
Transport of the incompletely spliced viral transcripts to the cytoplasm depends on an adequate supply of Rev [98]. Rev is a small shuttling protein that binds a complex RNA stem loop termed the Rev response element (RRE), which is located in the env gene. Rev binds first with high affinity to a small region of the RRE termed the stem loop IIB (Fig. 3.4) [101]. This binding leads to the multimerization of Rev on the remainder of the RRE. In addition to a nuclear localization signal, Rev contains a leucine-rich nuclear export sequence (NES) [98]. Of note, the study of Rev was the catalyst for the identification of such NESs in many cellular proteins and of the complex formed between CRM-1/exportin-1 and this sequence [98].
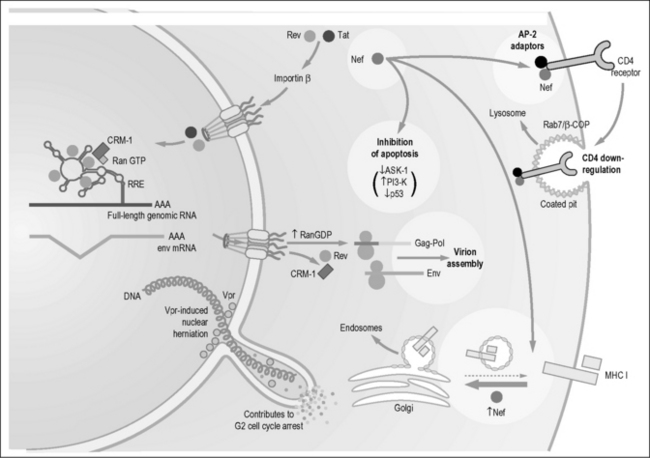
Rev functions by binding directly to CRM-1, the main nuclear export receptor for host ribosomal and small nuclear RNAs [102]. In addition to CRM-1, Rev also uses several other co-factors for RNA export. These factors include Rab/hRIP [103], RanBP1 [104], Sam68 [105], and heterogeneous nuclear ribonucleoprotein A1 (hnRNP A1). Ran is a small guanine nucleotide–binding protein that switches between GTP- and GDP-bound states. RanGDP is found predominantly in the cytoplasm because RanGAP is expressed in this cellular compartment. Conversely, the Ran nucleotide exchange factor RCC1, which charges Ran with GTP, is expressed predominantly in the nucleus. The inverse nucleocytoplasmic gradients of RanGTP and RanGDP produced by the subcellular localization of these enzymes likely plays a major role in determining the directional transport of proteins into and out of the nucleus. Outbound cargo is only effectively loaded onto the CRM-1/exportin-1 in the presence of RanGTP. However, when the complex reaches the cytoplasm, GTP is hydrolyzed to GDP and the bound cargo is released. The opposite relationship regulates the nuclear import by importins-α and -β, where nuclear RanGTP stimulates cargo release [98].
For HIV infection to spread, a balance between splicing and transport of incompletely spliced viral mRNA species must be achieved. If splicing is too efficient, then only the multiply spliced transcripts appear in the cytoplasm. Although required, these regulatory proteins are insufficient to support full viral replication. However, if splicing is markedly impaired, adequate synthesis of Tat, Rev, and Nef will not occur. In many non-primate cells, HIV transcripts may be overly spliced, thus producing a block to viral replication in these hosts [106].
The HIV-1 RNA transcript contains a long 5’-untranslated region (UTR) leader. Consequently, the start codons for Gag translation are often preceded by nonproductive start and stop codons. This situation suggests that HIV-1 might contain an internal ribosome entry site (IRES) to initiate translation [107]. Translation of the Gag–Pol polyprotein requires a ribosomal frameshift before the Gag stop codon and presumably host proteins to complete protein synthesis; however, the identities of these factors remain unknown.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


