Chapter 17 Medication problems and dialysis
Dialysis patients routinely take an average of 10 different medications and experience adverse drug reactions at least three times more frequently than the general population (Jick, 1977; St. Peter et al., 1997). To optimize drug outcomes, health professionals must be prepared to recognize and manage problems associated with medication use. Table 17-1 lists several problematic reactions to medications that clinicians encounter in providing care to patients on dialysis and to other patients with renal impairment.
Table 17-1 Problem Areas Involving Drugs
| Problem area | Corresponding responsibility of dialysis personnel |
|---|---|
| Drugs can damage kidneys, initiating or worsening renal failure. | Monitor renal function of patients on drugs or drug combinations that can damage kidneys. Identify patients at high risk for renal damage from drugs. Avoid or use extreme caution with drugs that damage renal function in high-risk patients and in those with existing renal disease. Initiate hydration and other documented measures to minimize nephrotoxicity. |
| Pharmacologic activity of drugs is altered by renal failure. | Adjust dosages to compensate for altered pharmacokinetic and pharmacodynamic activity. Monitor for therapeutic failure, adverse effects, or toxicity of all drugs used. Anticipate more adverse effects in patients with renal impairment. |
| The amount of medication removed from the body during dialysis varies, depending on the characteristics of the drug and dialysis conditions. | Using references and formulas, estimate how much drug is removed by dialysis. Calculate dosage adjustments and/or postdialysis replacement dosage. Monitor clinical response to calculated doses and alter the dosage as indicated. |
| Some poisons or drugs taken in overdose can be removed wholly or in part by dialysis. | Know which poisons and overdosed drugs can be removed by various dialysis procedures. Implement dialysis to treat poisoning and overdose, providing appropriate supportive care and observation during the procedure. |
| Medications may increase risks associated with the dialysis procedure. | Know what medications the patient is taking. Monitor for excess effects of the medication. |
How do drugs cause renal impairment?
Nephrotoxicity due to drugs contributes to 8% to 60% of AKI cases in hospitalized patients (Rosner & Okusa, 2008), The most commonly implicated pharmacologic nephrotoxins are antibiotics (aminoglycosides, cephalosporins, pentamidine, amphotericin B), radiocontrast agents used for radiologic studies, cyclosporine, cisplatin, angiotensin-converting enzyme (ACE) inhibitors, and nonsteroidal antiinflammatory drugs (NSAIDs). Because of the development of new agents (e.g., lower osmolar radiocontrast agents), changing drug use patterns (e.g., decreased use of aminoglycosides), and the shift of care from inpatient to outpatient settings, NSAIDs and ACE inhibitors are increasingly predominant causes of transient acute kidney injury. In chronic outpatient settings, chronic kidney disease (CKD) can occur due to combination analgesics, which consist of either aspirin or an NSAID combined with acetaminophen, caffeine, and/or codeine. Although the agents specifically cited here are the most frequent causes of renal damage, numerous other medications from diverse drug categories cause renal damage. The risk is greatest in individuals who already have poor kidney perfusion. Whenever a patient evidences renal impairment, a careful analysis of the drug profile for potential drug nephrotoxicity should be conducted. Nephrotoxins should be avoided in patients in any stage of CKD or used with appropriate dosage adjustments and meticulous monitoring.
How do these drugs usually cause renal damage?
Several mechanisms of renal damage by drug nephrotoxins have been identified (Rosner & Okusa, 2008), but most nephrotoxins damage the kidneys through more than one mechanism. Hemodynamic mechanisms involve inhibition of regulatory and compensatory processes, nonspecific renal vasoconstriction, or altered colloid oncotic pressure. A primary example of hemodynamic mechanisms for nephrotoxicity includes transient acute kidney injury from inhibition of the renin-angiotensin-aldosterone system by ACE inhibitors in patients with renal artery stenosis. A second example is the inhibition of prostaglandin-dependent renal blood flow by NSAIDs in patients with conditions associated with decreased renal blood flow (e.g., volume depletion and congestive heart failure). Renal vasoconstriction is a hypothesized mechanism of renal damage from propranolol, mannitol, a combination of triamterene and indomethacin, and the initial months of cyclosporine therapy. Dextran-40 can elevate oncotic pressure and impair glomerular filtration.
How can renal damage from such drugs be minimized or avoided?
Naturally, drugs with potential to cause renal damage should be avoided or used cautiously in patients with high risk for renal impairment. Conditions that predispose to renal damage by drugs include use of multiple nephrotoxins, sodium or fluid depletion, preexisting renal disease, and low renal blood flow in patients with diseases like congestive heart failure and cirrhosis, advanced age, and diabetes mellitus (Rosner & Okusa, 2008 ). Often drug-induced renal damage is reversible if the drug is discontinued and supportive care is initiated before permanent effects occur. Giving saline intravenously may decrease damage by some nephrotoxins such as cyclosporine and cisplatin by diluting the concentration of the drug in the renal tubule. Misoprostol, a prostaglandin analog, may prevent NSAID nephropathy. Drugs with the least nephrotoxic potential should be selected. For example, acetaminophen, aspirin, nonacetylated salicylates, sulindac, or nabumetone may have less nephrotoxicity than other NSAIDs. Finally, drugs should be given in the lowest effective doses for the shortest possible duration.
What are the pharmacokinetic parameters that reflect alterations caused by chronic kidney disease?
Each drug has a unique pattern of distribution throughout the body, represented as its apparent volume of distribution (Vd). Vd is the hypothetical volume that would be required to contain the dose of drug at its concentration in the plasma. For example, if 500 mg of a drug were administered to a patient and an hour later the concentration of the drug in a sample of that patient’s plasma was 0.001 mg/mL, the Vd would be 500 L. Stated another way, if each milliliter of plasma had 0.001 mg in it, 500 L would be required to contain 500 mg. Although Vd is an abstraction, it can be interpreted to reflect the distribution characteristics of a drug. If the Vd is 5 L (0.06 L/kg), it is likely that most of the drug stays in the intravascular space. If the Vd is more than 46 L (0.7 L/kg), the drug is sequestered in the peripheral tissues, usually dissolved in fatty tissues or bound to tissues. Drugs with large volumes of distribution are poorly dialyzable because most of the drug is outside the bloodstream and therefore not exposed to the dialysis membrane. Vd is used in the calculation of loading doses.
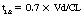
What factors affect absorption of medications in patients with chronic kidney disease?
Orally administered drugs pass through the liver before reaching the main circulation, and large portions of a dose of drugs like morphine, propranolol, and codeine are metabolized during this “first pass” through the liver. Because the first-past effect reduces the bioavailability (F) of orally administered morphine, the oral dose of morphine must be much larger than the injected dose to achieve the same amount of pain relief. In uremia, the fraction of drug metabolized by this first-pass effect may be decreased, unchanged, or increased because some metabolic by-products of uremia change the activity of liver enzymes. Thus bioavailability of drugs is highly variable in CKD.
What factors affect distribution of medications in patients with renal failure?
Plasma protein binding of acidic drugs to albumin may be decreased in renal failure as a result of either decreased concentration of albumin or decreased capacity of the albumin to bind to drugs. Changes in protein binding can alter Vd and the drug effect because only the free drug is pharmacologically active. Decreased albumin binding is thought to contribute to the central nervous system toxicity of acid drugs like theophylline, phenytoin, penicillin, phenobarbital, and salicylates in uremia. Some alkaline drugs (e.g., lidocaine, phenothiazines, propranolol, quinidine, and tricyclic antidepressants) that bind to glycoprotein also undergo increased or decreased binding in renal disease, but the clinical relevance of these changes is not as well studied as albumin binding. Although changes in Vd and plasma protein binding theoretically could have substantial effects on drug response, current research suggests that the effects of altered Vd in renal disease are usually minimal. There are a few exceptions, in which these changes require modification in the approach to patient care. An example is decreased protein binding of phenytoin during renal failure, which must be considered in clinical management. Measured serum concentrations of phenytoin that reflect total (bound + free) drug concentration in the plasma often are reported in the subtherapeutic range in patients with renal failure. This is because the amount of phenytoin bound to albumin is decreased, but the fraction of unbound drug is increased. Because the unbound drug is the active portion, lower drug concentrations of phenytoin are desirable for patients in renal failure to achieve the desired effect (Aweeka, 1995). In some centers both free and total phenytoin drug concentrations are measured to avoid toxicity. In patients without CKD, protein binding is about 90% of the total drug, whereas in patients with CKD binding of phenytoin ranges from 65% to 80%. Another approach to interpretation of phenytoin levels in CKD patients is to use a correction formula that adjusts the reported phenytoin plasma concentration for albumin level, renal function, and decreased affinity of phenytoin for albumin in CKD (Liponi et al., 1984).
What factors affect elimination of medications in patients with chronic kidney disease?
When a drug that is normally cleared unchanged by the kidneys is repeatedly administered to a patient with renal insufficiency, it begins to accumulate in the blood and may cause adverse effects. Increased portions of the drug may be eliminated by alternate routes, such as hepatic metabolism or through the lungs, bile, or sweat glands. Metabolites of drugs accumulate in renal insufficiency, and active or toxic metabolites contribute to adverse effects. An example is normeperidine, a metabolite of meperidine, which causes stupor or seizures when it accumulates. Box 17-1 includes examples of drugs with active or toxic metabolites that may accumulate in renal failure. If viable alternatives exist, drugs with active or toxic metabolites are avoided in patients with renal failure. When drugs with active or toxic metabolites are used in patients with renal failure, decreased dosages may be required and clinical monitoring must be vigilant. For example, patients with renal impairment who take allopurinol for gout or cancer require lower dosages than those with normal renal function because an active metabolite of allopurinol can cause exfoliative dermatitis when it accumulates in the body. Although far less important than active metabolites, inactive metabolites may also have consequences. For example, the accumulation of inactive metabolites may cause interference with laboratory tests.
How does dialysis affect pharmacokinetics of drugs and poisons?
The kidneys eliminate drugs through several processes. Although dialysis is not a substitute for all of these renal processes, some drugs are removed by dialysis. Dialysis may also affect other pharmacokinetic parameters. For example, changes in total body water from predialysis to postdialysis will affect the Vd of some drugs. Characteristics of drugs that promote removal by dialysis are as follows: (1) small molecular size, (2) small Vd, (3) water solubility, and (4) low protein binding. If protein binding exceeds 90%, the drug will be negligibly eliminated by dialysis. Drugs are more likely to be removed when the dialyzer membrane is highly permeable and its surface area is large and when the blood flow rate and dialysate flow rate are high. Peritoneal dialysis generally provides little drug removal because dialysate flow rate is slower than with other methods, although a greater amount of protein-bound drug can be removed due to large protein losses seen with this mode. Continuous therapy with hemofiltration or continuous hemodialysis for critically ill patients can remove substantial fractions of drugs. Removal of drugs by hemofiltration procedures is determined by the ultrafiltration rate and the degree of protein binding. Treatment of drug overdose and poisoning involves application of these principles to decrease serum concentration of the toxic drugs or substance (Winchester & Kriger, 1995). Similarly, poisons with high protein binding or large Vd are not dialyzable. Although many standard references classify a drug as dialyzable or not dialyzable, dialyzability is not an all-or-nothing characteristic. Some drugs are virtually entirely removed by dialysis; others have negligible removal. Many drugs fall somewhere in the middle. The type of dialysis equipment and the length of dialysis greatly influence whether a drug is removed. Classification of dialyzability as “yes” or “no” is based on an expert’s opinion of whether removal is clinically significant—that is, sufficient to remove an overdose—or whether the patient will require dosage replacement.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


