20 Management of Shock
Introduction
Shock is an altered physiological state that affects the functioning of every cell and organ system in the body. It is a complex syndrome reflecting changing blood flow to body tissues with accompanying cellular dysfunction and eventual organ failure.2,3 Shock presents as a result of impaired nutrient delivery to the tissue:
While the cause of shock may be multifactorial, treatment focuses on optimising tissue perfusion and oxygen de-livery. Shock is often classified according to the primary underlying mechanism: a disruption of intravascular blood volume, impaired vasomotor tone or altered cardiac contractility.5 The shock syndrome is one of the most pervasive manifestations of critical illness present in intensive care patients.
Early detection and management of shock to reverse pathological processes improves patient outcomes.6 Although the traditional hallmark of shock is hypotension (SBP <90 mmHg) this can be a late or misleading sign and is considered a medical emergency.7 It is therefore critical that other signs and symptoms are identified early by frequent observations to detect a patient’s deteriorating state and respond before irreversible shock ensues.8 No one vital sign is adequate in determining the level or extent of shock6 nor is there a specific laboratory test which diagnoses the shock syndrome.
Pathophysiology
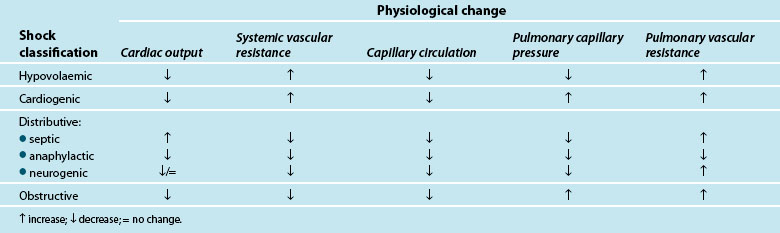
Traditionally, shock is classified by aetiology: hypovolaemic, cardiogenic and distributive.3,4,9 Each has a specific mechanism of action that leads to altered tissue perfusion and oxygen and nutrient uptake at the cellular level (see Table 20.1). In practice, it is common to find overlap between different shock types (e.g. in sepsis there may also be hypovolaemia and/or myocardial dysfunction).
| Shock type | Main characteristic |
|---|---|
| Hypovolaemic | a reduction in circulating blood volume through haemorrhage or dehydration or plasma fluid loss |
| Cardiogenic • obstructive shock | pump failure (impaired cardiac contractility) usually as result of myocardial infarction a sub category of cardiogenic shock characterised by blockage of circulation to the tissues by impedance of outflow or filling in the heart (e.g. due to cardiac tamponade or pulmonary emboli) |
| Distributive shock | a maldistribution of circulation from sepsis, anaphylaxis or neurogenic injury |
Shock occurs when there is an inability of the body to meet metabolic demands of the tissues; hypoperfusion (decreased blood flow to the tissues) results in cellular dysfunction, as there is homeostatic imbalance between nutrient supply and demand,4,10 and adaptive responses can no longer accommodate circulatory changes. These adaptive responses are moderated via numerous ‘sensors’ throughout the thorax and large vessels in particular, which detect subtle changes in pressure (baroreceptors) or biochemical changes (chemoreceptors). These receptors feed back to the hypothalamus which regulates through the pituitary gland (for the release of a number of hormones such as antidiuretic hormone [ADH] and adrenocorticoid trophic hormone [ACTH] to target organs such as the kidney) and the cortex of the adrenal gland to respond and counter the developing effects of shock. Concurrently direct feedback stimulates the sympathetic nervous system to act on blood vessel tone, particularly the arterioles, and also target organs such as the adrenal gland and kidney to respond via the release of endogenous catecholamines (adrenaline and noradrenaline), mineral and glucocorticoids (aldosterone, cortisol), and the renin–angiotensin–aldosterone system (RAAS). RAAS activation results in synthesis of angiotensin II, a powerful vasoconstrictor that further potentiates the reduction in peripheral blood vessel capacity.
As adaptive responses fail, cardiac output becomes insufficient to provide adequate organ perfusion despite increasing tissue oxygen consumption (see Chapters 9 and 10). When oxygen is ‘supply dependent’, oxygen delivery is decreased and, to compensate, increased extraction occurs to enable continued tissue consumption. However, when oxygen delivery falls below a critical threshold, and extraction demand rises above the available blood oxygen levels, this compensation mechanism fails and oxygen debt results.6,11,12
Hypoperfusion may also exist despite a relatively normal cardiac output, and may not be immediately evident clinically.6 This maldistribution of bloodflow to some tissues while other areas receive more blood flow than needed,4,7,10,13,14 is often referred to as distributive shock, and is typical of the shock types that affect vasomotor tone (e.g. septic, neurogenic and anaphylactic shock). This maldistribution may leave some organ systems ischaemic for long periods leading to persistent organ dysfunction and failure.6 There is also evidence supporting the presence of cytopathic hypoxia as a result of excessive nitric oxide and tumour necrosis factor-alpha (TNFα) production (cellular proinflammatory mediators), where there is impaired mitochondrial (the powerhouse of the cell) oxygen utilisation which leads to depleted stores of adenosine tri-phosphate (ATP)4,11,13,15,16 and interferes with electron transport and metabolism16 (see Chapter 19). Nitric oxide is associated with vascular relaxation and is a major contributor to alterations in microvasculature and capillary leak in sepsis.17
Organ systems have varying responses in shock and are not measured directly. Often surrogate markers of global hypoperfusion are used to indicate the severity of shock.18–19 Lactate and acid–base disturbances, such as an increase in strong ion gap, have been suggested as early markers of mitochondrial dysfunction and cellular hypoperfusion.8,20 These ‘surrogate’ biochemical markers of hypoperfusion (pH, serum lactate and standard base excess) assess acidaemia and provide some insight into the degree of shock present.21 Lactate, a strong anion with normal production of 1500–4500 mmol/day, is a product of carbohydrate metabolism. Increased levels are present in tissue hypoxia, hypermetabolism, decreased lactate clearance, inhibition of pyruvate dehydrogenase and activation of inflammatory cells; all characteristics of developing shock (see Table 20.2). Increased lactate production is a warning sign of impending organ failure, as it is indicative of anaerobic metabolism. Blood lactate levels have been directly linked to deteriorating patient outcomes in shock.21,22
TABLE 20.2 Lactate production9
| Lactate production | |
| Product of carbohydrate metabolism (1400–4500 mmol/day) | Glucose, glycolysis; pyruvate, lactate |
| Rise in lactate levels | |
| Tissue hypoxia | |
| Hypermetabolism | |
| Decreased clearance of lactate | |
| Inhibition of pyruvate dehydrogenase | |
| Activation of inflammatory cells | |
| Phagocytosis | |
| Major source in sepsis | |
| Phagocytes | |
As the shock state deteriorates and the body fails to compensate, organ systems begin to fail. This is complicated by a systemic inflammatory response (SIRS) which can be a direct cause of the shock state (see section on Distributive shock) or develop as a consequence of protracted shock. This results in ‘capillary leak’ or increased microvascular permeability which leads to interstitial oedema as a consequence of alterations to tissue endothelium. Many immune mediators including circulating cytokines, oxygen free-radicals and activated neutrophils alter the structure of the endothelial cells, creating space to allow larger intravascular molecules to cross into the extravascular space,23 with proteins and water moving from the intravascular space into the interstitium.24 This response mechanism improves the supply of nutrient-rich fluid to the site of local injury, however, systemically, fluid shifts lead to hypovolaemia, impaired organ function and development of acute organ injury such as acute lung injury (ALI) and acute kidney injury (AKI).24 This developing organ injury is the precedent to organ failure (more fully described in Chapter 21).
Patient Assessment
Critically ill patients often exhibit signs of tissue hypoxia as a result of cardiovascular disturbances.25 Table 20.3 provides an overview of the physiological changes in shock. Therapy is targeted to maintain oxygen delivery (DO2) to vital organs to prevent ischaemia and cell death.25,26 Ideally, organ systems and tissues should be monitored individually,25 however global measures such as perfusion pressure, cardiac output (CO) and DO2, are commonly used as surrogates to assist in treatment decision making.19 Patient assessment and haemodynamic monitoring, including calculation of CO, are used to differentiate shock states and assess progress in relation to treatment.26–28 CO is seen by many clinicians as an important assessment of shocked patients as it is a major determinant of DO2.25,26 Critically ill patients are frequently assessed clinically, although cardiac output estimations from physical examination are generally unreliable and patient status may change quickly.29 Therefore invasive techniques are most commonly used in critical care to measure CO (see also Chapter 9).
Non-Invasive Assessment
Perfusion status is determined clinically using gross organ function such as mental status, urine output and peripheral warmth and colour.6 Basic physical assessment of cardiovascular, central nervous system and renal function are essential when assessing a patient at risk of shock. Subtle changes in urine output, heart rate and capillary refill are all signs of physiological compensation in response to altered tissue perfusion associated with shock. Regular tracking of these vital signs and trend monitoring through careful documentation can alert clinicians to impending deterioration in the shock state. Level of consciousness may deteriorate; an early sign may be anxiety, and progress to restlessness, agitation or coma. Other assessment findings include cool, clammy skin, postural hypotension, tachycardia and decreased urine output.3 The reliability of these measures is questionable, particularly where multiple assessments by different clinicians are performed; in the ICU continuous ECG monitoring and invasive monitoring techniques are employed to assist in the objective assessment of changes in cardiovascular state.
Although CO estimations based on physical assessment findings are unreliable, physical examination using an estimation of vascular resistance has shown reasonable accuracy.30 Clinical assessment may determine CO using the rearranged equation of systemic vascular resistance (SVR = MAP − CVP/CO) where vascular resistance is measured through peripheral skin temperature changes.30 A reliable and accurate non-invasive clinical assessment technique of estimating cardiac output would be clinically useful27 allowing assessment of patients without invasive monitoring, or used to verify accuracy from invasive devices. While a number of non-invasive cardiac output measuring devices are available, further research and refinement is required before widespread application is considered in critical care.31
Invasive Assessment
The indicator dilution method using a thermal (thermodilution) signal (cold or hot) is the customary clinical standard for measuring CO26 in ICU. This is usually achieved by placement of a pulmonary artery catheter (PAC), or a central line in conjunction with a thermistor-tipped arterial cannula (transpulmonary aortic thermodilution). Other invasive techniques measure CO continuously using pulse contour or arterial pressure analysis and ultrasound doppler methods use an oesophageal probe. All methods have degrees of invasiveness, can be time-consuming to yield measurements of acceptable accuracy32, may be expensive and are not without risk of complications.27,33 The PAC is a controversial assessment tool26,28,33 due to the risk associated with the invasive line versus benefits for the measurement of CO34. This has led to increased interest in less or non-invasive measures of CO.
A further invasive assessment approach is the continuous estimation of mixed venous oxygen saturation using a light-emitting sensor in a PAC. As tissue oxygen delivery fails to meet demand and oxygen extraction rises, the residual oxygen content of blood returning to the lungs will fall; in effect a surrogate indicator of failure to meet body tissue oxygen demand. This technology was used in the landmark study by Rivers and colleagues.35 to monitor early deterioration of septic shock patients presenting to the ED in need of resuscitation and was part of a goal-directed approach to managing patients. This single-centre US study has been the subject of much interest for its claimed improvement in patient outcome, with this goal-directed approach being assessed in a major multicentre study in an effort to verify its findings within an international context and varying approach to critical care delivery.36
Management Principles
Managing a patient in shock focuses on treating the underlying cause, and restoration and optimisation of perfusion and oxygen delivery; this includes relevant activities using the acronym VIP37 (see Box 20.1). It is also suggested that giving critically ill patients a daily ‘FASTHUG’ improves the quality of care for patients in ICU.38 Specific management of patients with shock are discussed separately below depending on the cause.
Practice tip
Feeding (prevent malnutrition, promote adequate caloric intake)
Analgesia (reduce pain, improve physical and psychological wellbeing)
Sedation (titrate to the 3Cs – calm, cooperative, comfortable)
Thromboembolic prophylaxis (prevent DVT)
Head of bed elevated (up to 45° to reduce reflux and VAP)
Ulcer prophylaxis (to prevent stress ulceration)
Hypovolaemic Shock
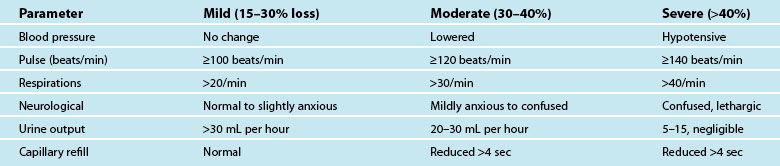
Hypovolaemia is a common primary cause of shock and also a factor in other shock states. Insufficient circulating blood volume is the underlying mechanism, leading to decreased cardiac output and altered perfusion.39,40 Death related to haemorrhage is most likely in the first few hours after injury.40 The most obvious cause is direct injury to vessels leading to haemorrhage, but there are more insidious causes such as dehydration from prolonged vomiting or diarrhoea, sepsis and burns.41 Hypovolaemic shock is classified as mild, moderate or severe, depending on the amount of volume loss (see Table 20.4). As the shock state worsens, associated compensatory mechanisms will be more pronounced,3 and hypovolaemic shock may deteriorate to Multi Organ Dysfunction Syndrome (MODS) if poor oxygen delivery is prolonged39 (see Chapter 21).
Clinical Manifestations
Symptoms of haemorrhage may not be present until more than 15–30% of blood volume is lost, and will deteriorate as the shock state worsens.3,41 Estimating blood or plasma loss is difficult and dilutional effects of resuscitation fluids may be evident when assessing haemoglobin and hematocrit.41 As the body compensates for the reduced circulating volume, widespread vasoconstriction occurs in most body systems apart from the heart and CNS; SVR rises markedly in an attempt to retain a viable circulatory system (this accounts for many of the signs and symptoms associated with circulatory compensation). However, as tissues are starved of oxygen and nutrients over a prolonged ischaemic time, local mediators are released as part of the inflammatory responses, leading to organ microvasculature vasodilation and capillaries re-open to maintain oxygen delivery and reduce hypoxia.41 This is a hallmark of developing MODS.
Nursing Practice
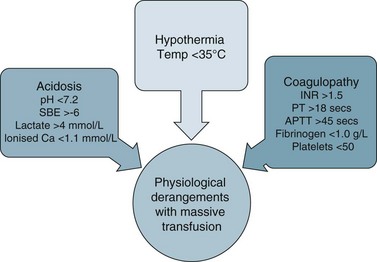
Clinical management of hypovolaemia centres on minimising fluid loss and rapid restoration of circulating blood volume41 once the airway and breathing are secure. More than one large-bore intravenous cannulae are usually inserted and lost circulating volume is replaced by colloids, isotonic crystalloids or blood products to achieve haemodynamic endpoints (e.g. MAP >65 mmHg). Body heat can be lost rapidly due to blood loss, the rapid infusion of room temperature fluids and exposure in the pre-hospital setting or during repeated physical examination. It is therefore important to institute measures to maintain patient temperature >35°C to avoid coagulopathies and loss of thermoregulation.42 The aim is to ameliorate the lethal triad of anaemia, coagulopathy and hypothermia.40–42
Debate surrounds early surgical intervention prior to aggressive fluid resuscitation.40 The premise is that allowing a lower perfusion pressure prior to achieving haemostasis with controlled or no fluid infusion results in less blood loss, due to the compensatory mechanisms described above.40 Use of medications such as Factor IVa and EPO also remains controversial in the setting of critical haemorrhage.42 Guidelines for ‘massive transfusion’ currently being finalised by the National Blood Authority (NBA) do not recommend use of Factor IVa beyond licensed indications, although there may be an indication when conventional therapy has failed to secure haemostasis following massive blood loss and transfusion of blood products. The current debate also includes dosage and thromboembolic complications associated with its use.42 The NBA is a statutory agency established in 2003 to improve and enhance the management of the Australian blood banks and plasma product sector. Current initiatives of the agency include development and promulgation of evidence based guidelines for both massive transfusion and intensive care.
Fluid resuscitation
Fluid resuscitation is a first-line treatment for hypovolaemic shock; providing fluid volume increases preload and therefore cardiac output (Starling’s law) and organ perfusion. A related principle is that the fluid infused should reflect fluid loss, e.g. plasma replacement in burns, fresh blood in massive haemorrhage. Giving a ‘fluid challenge’ is not always appropriate; the determining factors will be assessment of volume responsiveness, and whether the infusion will not be deleterious, causing overload, fluid shifts and perpetuating inflammatory responses.6 The fluid type, volume, rate and targeted endpoints is documented;41 often this is structured as a bolus dose in volume/kg to achieve a measured haemodynamic variable. When massive transfusion is required, attention should be given to product selection and hence a protocol can be employed.
Independent Practice
Critical care nurses must be efficient and practised at initial patient assessment to establish the degree of compensation occurring in a hypovolaemic patient. Figure 20.1 highlights clinical manifestations of haemorrhage. Careful consideration of a patient’s clinical picture will establish a hierarchy and priority of needs. Most hospitals have some level of track and trigger response that escalates care to appropriate levels (e.g. MET calling criteria), however nurses are in a position to establish first line management such as intravenous access where this is a required skill. There are also many examples of protocols and guidelines for nurses to initiate fluid resuscitation where a patient has indications of inadequate circulating blood volume; e.g. a fluid bolus up to 20 mL/kg of colloidor 30–40 mL/kg crystalloid may be recommended (depending on organisational guidelines).
Collaborative Management
Selection of the appropriate fluid indications for surgical management and ‘permissive hypotension’ (deliberate limiting or minimising resuscitation until after adequate surgical control of haemorrhage).40,42 will be assessed by the multidisciplinary team. Goal-directed therapy includes prevention of tissue hypoxia, typically through rigorous fluid resuscitation with either crystalloids or colloids to achieve specific haemodynamic endpoints (e.g. a CVP of 8–12 mmHg, MAP >70 mmHg, urine output >0.5 mL/kg/h). Vasopressor and inotrope therapy may be then added to maintain adequate perfusion pressure; noradrenaline is the vasopressor of choice because of vasoconstrictor effects.43
Preload management
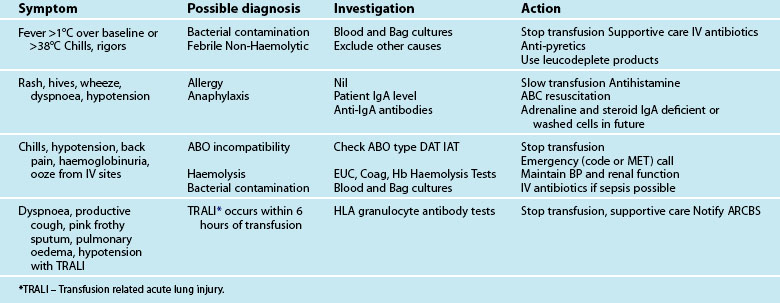
The colloid versus crystalloid fluid resuscitation debate (use of albumin-based solutions or colloids) continues despite findings from the SAFE study conducted in Australasia; crystalloids (isotonic saline based solutions) were as effective as colloids for fluid resuscitation.44–46 The scientific rationale for using colloids over crystalloids is to preserve plasma oncotic pressure so as to retain intravascular fluid and minimise oedema. Colloids may also attenuate the inflammatory response.20 If moderate to severe hypovolaemia is suspected then blood is often used to improve oxygen-carrying capacity. Further dilution of blood by volume expanders increases hypoxia (otherwise known as isovolaemic anaemia) and red cells are usually needed. Use of isotonic saline as a volume expander is common, although resuscitation with large volumes of saline solutions can be associated with hyperchloraemic acidosis.40 Blood and blood components are usually considered necessary where patients exhibit signs of moderate to severe haemorrhage (see Figure 20.2). There is no perfect resuscitation fluid, and selection is guided by patient condition and the type of fluid lost.
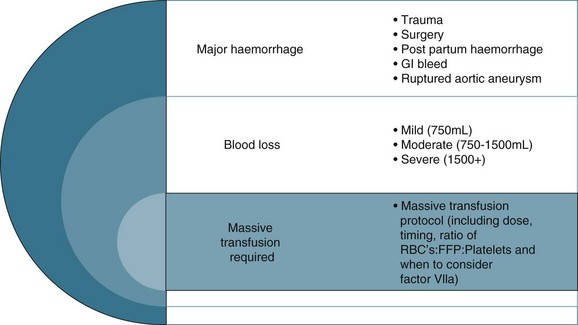
There are a number of factors to consider when administering blood products in massive volume. Massive transfusion is defined as replacement of a patient’s total blood volume in less than 24 hours (approximately 10 units of red cells);47,48 although the literature is inconsistent.48 A number of complications are evident (e.g. transfusion reactions, coagulopathies, hypothermia, sepsis)3 and is associated with high mortality.48 Patients receiving massive blood transfusions require careful monitoring for signs of metabolic derangements, hypothermia, citrate toxicity, hyperkalaemia and coagulopathies (due to depletion of clotting factors). Dilution and clotting factor consumption cause microvascular bleeding, often manifesting as oozing from multiple sites even after surgical correction.47,48 Massive transfusion of stored blood with high oxygen affinity adversely affects oxygen delivery to the tissues. It is therefore preferable to transfuse blood cells that are less than 1 week old; 2,3-diphosphoglycerate levels rise rapidly after transfusion, and normal oxygen affinity is usually restored within a few hours of transfusion.47
Each unit of blood contains approximately 3 g of citrate, which binds to ionised calcium. A healthy adult liver metabolises 3 g of citrate every 5 minutes. If blood is transfused rapidly or the liver is impaired, citrate toxicity and hypocalcaemia may develop. The patient should therefore be monitored for signs of tetany, hypotension and electrocardiographic evidence of hypocalcaemia.47 As stored blood ages, plasma potassium levels rise (possibly to over 30 mmol/L). Hypokalaemia may be more common as red cells begin active metabolism and intracellular uptake of potassium restarts with transfusion.47
Acid–base disturbances may also be evident due to the stored blood lactic acid levels and the citric acid. Citrate metabolises to bicarbonate, and a profound metabolic alkalosis may result from massive blood transfusion. As hypothermia causes reduced citrate and lactic acid metabolism, an increase in the affinity of haemoglobin to oxygen, platelet dysfunction and an increased tendency for cardiac dysrhythmias,47 the patient and the blood transfused should be warmed to avoid complications.
Leucocyte depletion occurs during donation in Australia and decreases up-regulation of the inflammatory immune response associated with transfusion. Current clinical practice guidelines for the administration of blood products and red cells to stable adult patients are listed in Tables 20.5 and 20.6. A new structure with multiple guidelines has been developed and the massive transfusion guideline is complete and will be followed by a number of other specialised guidelines. All guidelines will be available to download from the National Blood Authority website as they are completed (see Online resources).
TABLE 20.5 Clinical practice guidelines for red blood cell and platelet administration
| Appropriate Use of Blood Components For Stable Adults & Children >4 months (corrected) age Adapted from NHMRC/ASBT guidelines (www.anzsbt.org.au) Haemoglobin is NOT the sole deciding factor for transfusion – consider other patient factors e.g. signs of hypoxia and ongoing blood loss. | |
| Red Cells | |
| Hb | Considerations |
| <70 g/L | Transfusion is often clinically useful unless early Hb recovery is expected. A threshold of <60 g/L may be appropriate for children. |
| 70–100 g/L | Likely to be appropriate during surgery with major blood loss or if there are signs or symptoms of impaired oxygen transport. |
| >80 g/L | May be appropriate to control anaemia-related symptoms in a patient on a chronic transfusion regimen or during marrow suppressive therapy. |
| >100 g/L | Not likely to be appropriate unless there are specific indications |
| WHAT DOSE? Red Cells (mL) = 0.4 × wt (kg) × (desired – actual) Hb (g/L) | |
| Platelets | |
| Use of platelets is likely to be appropriate as prophylaxis for: | |
| Indication | Considerations |
| Bone Marrow Failure | At a platelet count of <10 Ö 109/L in the absence of risk factors and <20 Ö 109/L in the presence of risk factors (e.g. fever, antibiotics, evidence of haemostatic failure) |
| Surgery/Invasive | To maintain platelet count at >40 Ö 109/L. For surgical procedures with high risk of bleeding (e.g. ocular or neurosurgery) it may be procedure appropriate to maintain at 100 Ö 109/L |
| Platelet Function Disorders | May be appropriate in inherited or acquired disorders, depending on clinical features and setting. In this situation, platelet count is not a reliable indicator |
| Use of platelets is likely to be appropriate as therapy for: | |
| Bleeding | Any patient in whom thrombocytopenia is considered a major contributory factor. |
| Massive Bleeding/Transfusion | Confined to patients with thrombocytopenia and/or functional abnormalities who have significant bleeding. Often with platelet count <50 Ö 109/L (<100 Ö 109/L with diffuse microvascular bleeding). |
Cardiogenic Shock
Cardiogenic shock manifests as circulatory failure from cardiac dysfunction,49 and is reflected in a low cardiac output (CI <2.1 L/min/m2), hypotension (SBP <90 mmHg) and severe pulmonary congestion, high central vascular filling pressures (CVP; PAOP >18 mmHg).50 Additional invasive parameters are: intrathoracic blood volume index >850 mL/m2; global end-diastolic volume >700 mL/m2; and extravascular lung volume index >10 mL/kg.51,52 Cardiogenic shock is commonly associated with AMI and manifests when 40% or more of the left ventricle is ischaemic. It is also related to mechanical disorders (e.g. acute cardiac valvular dysfunction or septal defects), deteriorating cardiomyopathies or congestive cardiac failure,53,54 trauma and obstruction or inhibition of left ventricular ejection (referred to as obstructive shock e.g. pulmonary emboli, dissecting aneurysm, tamponade)37,53 (see Chapter 10). Myocardial depression from non-cardiac causes such as sepsis, acidosis, myocardial depressant factor, hypocalcaemia or drug impact55 may be so severe as to present as cardiogenic shock.
Incidence has been estimated at 3% of patients presenting with AMI, and mortality remains high (50–80%),56 given death from AMI overall is 7%. This is despite treatment advances including emergency revascularisation.57,58 Wider distribution of interventional cardiac revascularisation services has likely improved outcome for patients who present early in the course of their acute disease.
Clinical signs include poor peripheral perfusion, tachycardia and other signs of organ dysfunction such as confusion, agitation, oliguria, cool extremities, dyspnoea, many of which are present in hypovolaemic shock.49 Compensatory mechanisms are conflicting for a patient with cardiogenic shock, as cardiac workload is increased on an already-failing heart yet cardiac muscle oxygen delivery may be compromised.54 A careful but rapid assessment of the clinical history is helpful in differentiating the precipitant cause of this shock.
Clinical Manifestations
The clinical features of cardiogenic shock are reflective of congestive cardiac failure, although with greater severity:50,51,58
• low cardiac output and hypotension
• poor peripheral perfusion: pale, cool, clammy peripheries
• altered mentation, restlessness and anxiety
• pulmonary congestion with widespread inspiratory crackles and hypoxaemia (perhaps with frank pulmonary oedema)
• respiratory alkalosis (hyperventilation) or acidosis (respiratory fatigue)
In the absence of invasive monitoring, the profile of hypotension, peripheral hypoperfusion, and severe pulmonary and venous congestion are evident although this ‘classic’ profile is not universal. On initial examination, 30% of patients with shock of left ventricular aetiology had no pulmonary congestion and 9% had no hypoperfusion.59
Based on the underlying pathology of an acute left ventricular myocardial infarction, the structural or contractile abnormality impairs systolic performance resulting in incomplete left ventricular emptying.50 This results in subsequent progressive congestion of first the left atrium, then the pulmonary circulation, right ventricle, right atrium and finally the venous circulation.50,60,61 When invasive haemodynamic monitoring is available, sequence of changes exist as illustrated in Figure 20.3.
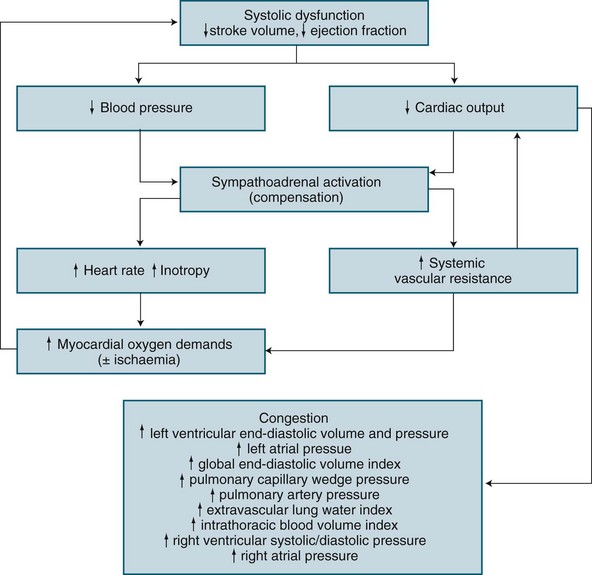
A patient with cardiogenic shock is also assessed and monitored for their oxygen delivery and tissue oxygen requirements (oxygen consumption). Systemic DO2 falls in proportion to a declining cardiac output, and is further worsened as hypoxaemia develops due to pulmonary oedema. Initially, VO2 may be sustained by an increase in tissue oxygen extraction ratio (O2ER).62 Normally 25% of delivered oxygen is extracted by tissues, but as delivery falls, tissues extract proportionally more oxygen to meet metabolic needs. Oxygen consumption can therefore be sustained until the severity of oxygen delivery deficit exceeds the ability to increase extraction. Maximal extraction is approximately 50%, and consumption falters when oxygen delivery falls to around 500–600 mL/min (cardiac index <2.2 L/min/m2).62–64 While use of a PAC is a well-described measure of severity in cardiogenic shock (as with hypovolaemic shock), evidence of improved patient outcome is unclear.28,65
Once oxygen consumption falls below tissue needs, resulting anaerobic metabolism causes lactate generation and the subsequent lactic acidosis.50,62 Progressive tissue ischaemia and injury ensues, along with worsening metabolic acidosis unless oxygen delivery can be restored. Myocardial contractile performance further worsens when myocardial ischaemia develops or when existing ischaemia or infarction is worsened, and a vicious cycle of ischaemia and dysfunction ensues.62
• Tachycardia offsets low stroke volume but increases myocardial oxygen consumption and decreases diastolic duration, reducing coronary perfusion time.
• Vasoconstriction limits the severity of hypotension but increases resistance to left ventricular emptying and may contribute to worsening of the cardiac output, in particular when cardiogenic shock is due to contractile dysfunction.
• An increase in cardiac workload to overcome the rise in systemic afterload increases myocardial oxygen demand, but cannot be met due to coronary artery occlusion.
• Developing pulmonary congestion is no longer contained within the pulmonary capillary and moves into the alveolar capillary space, creating pulmonary oedema, further impeding oxygen delivery to the circulation.
Nursing Practice
Independent Practice
Assessment
Frequent, thorough assessment of the patient’s status is essential, focusing on:
1. identification of patients at risk of clinical deterioration;
2. assessment of the severity of shock and identification of organ or system dysfunction;
3. assessment of the impact of treatment; and
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


