Chapter 27 Acute kidney injury (AKI) is a relatively new term used to describe the spectrum of acute-onset kidney disorders that can range from mild impairment of kidney function through acute kidney failure that requires renal replacement therapy (dialysis).1,2 Severe AKI is characterized by a sudden decline in glomerular filtration rate (GFR), with subsequent retention of products in the blood that are normally excreted by the kidneys; this disrupts electrolyte balance, acid–base homeostasis, and fluid volume equilibrium. A transition to greater use of the word kidney rather than renal reflects a trend in the nephrology literature that emphasizes the vulnerability of the kidney during critical illness.1,2 Critical care patients with AKI have a longer length of hospital stay, more complications and higher mortality.1,2 AKI-associated mortality ranges from 15% to 60%.2 One of the reasons for poor survival is that critical care patients often have coexisting nonkidney conditions that increase their susceptibility to the development of AKI. High-risk diagnoses include heart failure, shock, respiratory failure, and sepsis. An observational study that examined the incidence and course of severe AKI in 618 patients in six academic medical centers in the United States found that AKI was accompanied by multiorgan failure in most patients, even those who did not require dialysis.3 In this observational study 64% of patients required dialysis, the in-hospital mortality rate was 37%, and the rate of nonrecovery of kidney function (permanent dialysis) or death was 50%.3 The mortality rate exceeded 50% when four or more body systems had failed.3 Other clinical studies report similar findings with equally high mortality.2 Today the most frequent causes of AKI in the critically ill are associated with sepsis and cardiac surgery.1 One of the challenges of estimating the incidence of AKI in the critical care unit has been the wide variation in definitions that have been used. Measurement of kidney function is indirect, and the diagnosis of AKI is predominantly derived from changes in urine output and elevation of serum creatinine level, with the understanding that changes in these values reflect a decline in the GFR.1,2 Urine output is sometimes a problematic measure to use because diuretics artificially increase the urine output but may not alter the course of kidney failure. The clinical insult may have direct effects on the kidney, such as the inflammation associated with sepsis, which accounts for almost 50% of the AKI seen in critical care units.1 The risk of critically ill patients developing AKI has been classified by a multinational group of nephrologists.1,2 The classification uses the acronym RIFLE—risk, injury, failure, loss, and end-stage kidney disease (ESKD).2 The RIFLE system classifies AKI in three categories of increasing severity (R, I, F) and two outcome criteria (L, E) based on GFR status reflected by the change in urine output or loss of kidney function2 (Table 27-1). If AKI is superimposed on a kidney that is already compromised, the term chronic is added to the RIFLE criteria to denote the cause as acute-on-chronic kidney failure.2 TABLE 27-1 RIFLE CRITERIA FOR ACUTE KIDNEY DYSFUNCTION AKI, Acute kidney injury; Cr, creatinine; UO, urine output. *All serum creatinine references are based on changes from baseline. Data from Kellum JA, et al. Definition and classification of acute kidney injury. Nephron Clin Pract. 2008;109(4):c182. The Acute Kidney Injury Network (AKIN) criteria are listed in Box 27-1. These criteria are similar to those proposed by the RIFLE group, and both groups intend to make the point that in the acutely ill patient, small changes in the serum creatinine level and urine output may signal important declines in the GFR and kidney function. A conceptual model that combines the features of the RIFLE criteria and AKIN criteria is shown in Figure 27-1.2 Previously, ARF was predominantly classified by the location of the insult relative to the kidney: prerenal (before), intrarenal (within), and postrenal (after) (Box 27-2). This remains a useful way to imagine the relationship between anatomy and functional insults to the kidney, although it is not evident that insults classified in this manner have any impact on outcomes of patients with AKI. Any condition that decreases blood flow, blood pressure, or kidney perfusion before arterial blood reaches the renal artery that supplies the kidney may be anatomically described as prerenal AKI.1 When arterial hypoperfusion due to low cardiac output, hemorrhage, vasodilation, thrombosis, or other cause reduces the blood flow to the kidney, glomerular filtration decreases, and consequently, urine output decreases (see Box 27-2). This is a major reason the critical care nurse monitors the urine output on an hourly basis.4,5 Initially, in prerenal states, the integrity of the kidney’s nephron structure and function may be preserved. If normal perfusion and cardiac output are restored quickly, the kidney will recover and not suffer permanent injury. However, if the prerenal insult is not corrected, the GFR will decline, the blood urea nitrogen (BUN) concentration will rise (prerenal azotemia),1 and the patient will develop oliguria and be at risk for significant kidney damage. Oliguria, defined as urine output less than 400 mL/day, or urine output less than 0.5 mL/kg/hr,4 with an elevated serum creatinine, is a classic finding in AKI. Prerenal azotemia is associated with a lower mortality than other forms of AKI.6 Any condition that produces an ischemic or toxic insult directly at parenchymal nephron tissue places the patient at risk for development of intrarenal AKI1 (see Box 27-2). Ischemic damage may be caused by prolonged hypotension or low cardiac output. Toxic injury reaction may occur in response to substances that damage the kidney tubular endothelium, such as some antimicrobial medications and the contrast dye used in radiologic diagnostic studies. The insult may involve the glomeruli and the tubular epithelium. When the internal filtering structures are pathologically affected, the condition was previously known as acute tubular necrosis (ATN), although the newer term of AKI is now more often used.1 Any obstruction that hinders the flow of urine from beyond the kidney through the remainder of the urinary tract may lead to postrenal AKI. This is not a common cause of kidney failure in the critically ill.1 When monitoring of the urine output reveals a sudden decrease in the patient’s urine output from the urinary catheter, a blockage may be responsible. Sudden development of anuria (urine output less than 100 mL/24 hr) should prompt verification that the urinary catheter is not occluded. After AKI is suspected, the degree of injury is assessed using blood analysis. Most serum levels of electrolytes become increasingly elevated as AKI develops (Table 27-2). Urinalysis values are also altered by AKI although these values are not predictive of outcome in critical illness. Consequently urinary electrolytes are rarely measured1 (Table 27-3). Clinical findings associated with AKI are listed in Table 27-4. Normal and abnormal urinalysis findings and reasons for their significance are summarized in Chapter 26. TABLE 27-2 NORMAL SERUM ELECTROLYTE VALUES TABLE 27-3 INITIAL URINE LABORATORY ANALYSIS FINDINGS IN ACUTE KIDNEY INJURY* *Results of urine laboratory tests are valid only in the absence of diuretics. †Urine in prerenal failure is concentrated, with low sodium. ‡Urine in intrarenal failure shows kidney damage because the nephron cannot concentrate urine or conserve sodium, and evidence of kidney damage (casts) is seen. §Urine test results in postrenal failure vary because the findings initially depend on the hydration status of the patient rather than the status of the kidney. TABLE 27-4 SERUM ELECTROLYTES IN ACUTE KIDNEY FAILURE Acidosis (pH less than 7.35) is one of the trademarks of severe acute kidney insult. Metabolic acidosis occurs as a result of the accumulation of unexcreted waste products. The acid waste products consist of strong negative ions (anions), elevated serum phosphorus levels (hyperphosphatemia), and other normally unmeasured ions (e.g., sulfate, urate, lactate) that decrease the serum pH. A low serum albumin concentration, which often occurs in AKI, has a slight alkalinizing effect, but it is not enough to offset the metabolic acidosis. Even respiratory compensation and mechanical ventilatory support are rarely sufficient to reverse the metabolic acidosis. Acidosis in AKI is complex, as evidenced by the fact that many AKI patients maintain a normal anion gap. The reasons for this remain unknown. Information on acidosis and arterial blood gas interpretation is presented in Chapter 19. Anion gap measurement is discussed in Chapter 26. The BUN level is not a reliable indicator of kidney injury as an individual test.1 The BUN concentration is changed by protein intake, blood in the gastrointestinal tract, and cell catabolism, and it is diluted by fluid administration. A BUN-to-creatinine ratio may be calculated to determine the cause of the AKI (see Table 27-3). The BUN-to-creatinine ratio is most useful in diagnosing prerenal AKI (often described as prerenal azotemia), in which the BUN level is greatly elevated relative to the serum creatinine value.6 Creatinine is a byproduct of muscle metabolism that is formed nonenzymatically from creatine in muscles.7 Creatinine is completely excreted when kidney function is normal.7 Consequently, when the kidneys are not working, the serum creatinine level rises. Even small increases in serum creatinine represent a significant decrease in GFR.7 Serum creatinine level is assessed daily to follow the trend of kidney function and to determine whether it is stable, getting better, or getting worse. If the patient is making sufficient urine, the urinary creatinine clearance can be measured. A normal urinary creatinine clearance rate is 120 mL/min, but this value decreases with kidney failure. Critical care patients with severe AKI will manifest elevated serum creatinine and may be oliguric. Consequently, the urinary creatinine clearance rate is rarely measured during critical illness.7 The fractional excretion of sodium (FENa) in the urine can be measured early in the AKI course to differentiate between prerenal AKI and inter-renal AKI (parenchymal). A FENa value below 1% (in the absence of diuretics) suggests prerenal compromise, because resorption of almost all the filtered sodium is an appropriate response to decreased perfusion to the kidneys. If diuretics are administered, the test is meaningless. A FENa value above 2% implies the kidney cannot concentrate the sodium and that the damage is intrarenal (ATN). FENa values do not have any predictive benefit in critical illness and are rarely measured.1 Urinary sodium is measured in milliequivalents per liter (mEq/L). The interpretation of results is similar to the FENa. A urinary sodium concentration less than 10 mEq/L (low) suggests a prerenal condition. A urinary sodium level greater than 40 mEq/L (in the presence of an elevated serum creatinine and the absence of a high sodium load) suggests intrarenal damage has occurred (see Table 27-2). As with other urinalysis tests, the use of diuretics invalidates any results. Because the diuretics alter resorption of water and produce dilute urine, the test result will not reflect actual kidney function. Many patients come into the critical care unit with disease states that predispose them to the development of AKI. Many others already have kidney damage but are unaware of this condition (Fig. 27-2). The incidence of chronic kidney disease (CKD) in United States is about 10%, or 1 in 10 people.8 Clinical practice guidelines for the management of ESKD categorize kidney dysfunction into five stages. Because of the large numbers of adults with kidney dysfunction (diagnosed or not), kidney function must be assessed on all critically ill patients at risk for fluid and electrolyte imbalance. The GFR associated with each stage and the numeric population estimates for each stage of kidney dysfunction are shown in Table 27-5. TABLE 27-5 DECREASED KIDNEY FUNCTION BY STAGE IN ADULT U.S. POPULATION *Data from Coresh J, et al. Prevalence of chronic kidney disease and decreased kidney function in the adult US population: Third National Health and Nutrition Examination Survey. Am J Kidney Dis. 2003;41(1):1. †Data from Nickolas TL, et al. Awareness of kidney disease in the US population: findings from the National Health and Nutrition Examination Survey (NHANES) 1999 to 2000. Am J Kidney Dis. 2004;44(2):185. Most people in the early stages of kidney disease are unaware of their condition.9 A national health survey queried individuals about whether they had ever been told by their physician that they had “weak or failing kidneys.” The answer to this question was correlated with the individual’s GFR and the presence of albuminuria by urine test to stratify them according to the five stages of CKD (see Table 27-5). The results showed that more than one half of the respondents were unaware that they had kidney dysfunction until they reached stage 5 or ESKD, when they would become dialysis dependent.9 In contrast, the prevalence of CKD in individuals between 18 and 39 years of age is only 0.5%.8 CKD results, categorized by stage of CKD, are listed in Table 27-5. There is a strong association between kidney failure and heart failure.11 In studies of critically ill patients with AKI, 54%3 have acute kidney failure and heart failure. Not all patients with kidney failure and heart failure have the same pathology, and this variation has been recognized. Heart-kidney interactions have been categorized into five subtypes under the term cardio-renal syndromes.12 The purpose of the new classification system is to identify relevant biomarkers, treatments, and future avenues for research.12 There is a significant association between respiratory failure and kidney failure. In studies of critically ill patients with kidney failure, over 50% have respiratory failure.3 Mechanical ventilation can alter kidney function. Positive-pressure ventilation reduces blood flow to the kidney, lowers the GFR, and decreases urine output.13 These effects are intensified with the addition of positive end-expiratory pressure (PEEP).13 AKI increases inflammation, causes the lung vasculature to become more permeable, and contributes to the development of acute respiratory distress syndrome (ARDS). Patients with AKI are more likely to require prolonged mechanical ventilatory support.14,15 Sepsis causes almost half of the cases of AKI in the critically ill.1 Sepsis and septic shock create hemodynamic instability and reduce perfusion to the kidney. Immunologic, toxic, and inflammatory factors may alter the function of the kidney microvasculature and tubular cells. Clinical guidelines for hemodynamic support in sepsis emphasize the need for adequate fluid resuscitation, because reversal of hypotension and restoration of hemodynamic stability can often be achieved with fluids alone.16 Unfortunately, in severely septic patients, inflammation increases vascular permeability, and much of this fluid may move into the third space (interstitial space). If the blood pressure remains low, the use of vasopressors is recommended to raise refractory low blood pressure after volume resuscitation.16 Vasopressors raise blood pressure and increase systemic vascular resistance (SVR), but they also may raise the vascular resistance within the kidney microvasculature. Other practices aimed at reversing the deleterious effects of sepsis include maintaining the patient’s hemoglobin level at 7 to 9 g/dL and blood glucose level below 150 mg/dL and ensuring optimal hydration as evidenced by a central venous pressure above 8 mm Hg.16 Trauma patients have different demographics from those of other critical care populations. They are always emergency admissions, are younger, are more often male, and have fewer coexisting illnesses.17 A 5-year retrospective study of 9449 trauma admissions to critical care units in Australia and New Zealand used the RIFLE criteria to determine incidence of AKI in the first 24 hours after admission; 18% of trauma patients developed AKI.17 However, if patients were older or had pre-existing comorbid illnesses, their risk of AKI rose to 35%.17 Although these AKI numbers are high, they likely underestimate the true incidence because the study did not include patients who developed AKI later than 24 hours after admission to the critical care unit.17 Trauma patients with major crush injuries have an elevated risk of kidney failure because of the release of creatine and myoglobin from damaged muscle cells, a condition called rhabdomyolysis.18 Myoglobin in large quantities is toxic to the kidney. A major goal of treatment is to prevent rhabdomyolysis-induced AKI. Mortality rate is low, provided adequate crystalloid volume is administered early in the course of treatment.19 It is important to trend the serum potassium levels. Life-threatening hyperkalemia can occur as cell lysis permits intracellular potassium to be released into the bloodstream.20 The level of creatine kinase (CK), a marker of systemic muscle damage, increases in patients with rhabdomyolysis. One trauma service reported that of 2083 critical care trauma admissions, 85% had elevated CK levels, and 10% developed AKI resulting from rhabdomyolysis.21 A CK level of 5000 units/L was the lowest abnormal value in patients who developed AKI associated with rhabdomyolysis.21 Crystalloid volume resuscitation is the primary treatment for preservation of adequate kidney function and prevention of AKI. In many hospitals, the intravenous (IV) fluids are alkalinized by the addition of sodium bicarbonate, and the urine output is increased by IV administration of the diuretic mannitol. A bicarbonate and mannitol regimen is instituted to prevent acidosis and hyperkalemia, because both are frequent complications of rhabdomyolysis. Close attention is paid to hourly urine output that can be dark brown or tea-colored,19 CK levels, serum creatinine levels, serum potassium levels, and any signs of compartment syndrome in all patients admitted with this diagnosis. More than 1 million radiologic studies or procedures that involve use of IV radiopaque contrast are performed every year. Approximately 1% of those patients will require dialysis as a result of contrast-induced nephrotoxicity (CIN).22 Patients at highest risk are those with pre-existing CKD, baseline serum creatinine levels more than 1.5 mg/dL, dehydration, diabetes, heart failure, or advanced age (older than 75 years).23,24 The clinical definition of CIN is an increase in serum creatinine concentration of 0.5 mg/dL or more, or a 25% increase from the patient’s baseline within 3 days of contrast medium exposure, without an alternative clinical explanation for development of AKI.23 The effects of reversible, contrast-induced AKI may not be limited to the immediate hospitalization; it has been linked to increased mortality in the 5-year period after the reversible AKI, compared with similar patients who did not have kidney injury.25 The best method of prevention is aggressive hydration with IV normal saline during and after the procedure.23 After some diagnostic intravascular catheterization procedures, the alert patient is asked to drink several liters of water over a 12-hour period to protect the kidney. Avoiding dehydration is vital. The addition of sodium bicarbonate, because of its alkalinizing effect, may confer protection to the vulnerable kidney beyond hydration with normal saline only.23 Other strategies such as adjunctive use of N-acetylcysteine or hemofiltration have shown conflicting results in research studies and are not recommended.23 Potentially nephrotoxic medications are also stopped before the procedure. Metformin, a medication that decreases insulin resistance in type 2 diabetes, has been associated with lactic acidosis in rare instances.23 For patients with elevated serum creatinine, Metformin is stopped the day before any procedure involving contrast and not started again for 48 hours when serum creatinine has returned to baseline.24 The majority of critically ill patients have a urinary drainage catheter inserted to accurately record urine output.26 This is an appropriate use of a urinary catheter. However, because of the risk of catheter-associated urinary tract infection (CAUTI), the catheter should be removed as soon as clinically feasible.27 Critically ill patients who have a protracted illness have a significant risk of contracting a CAUTI, especially if the catheter is required for several days.28 Additionally, critically ill patients who have developed a CAUTI have a higher mortality and longer lengths of stay.29 The focus on prevention of CAUTI has gained considerable momentum. The Centers for Medicare and Medicaid Services (CMS) will no longer provide reimbursement for hospital-acquired CAUTI because it is considered a preventable infection. The Joint Commission has added prevention of CAUTI as a 2012 patient safety goal.30 Currently only 40% of hospitals routinely collect data on the rates of CAUTI in critical care units as part of a prevention and monitoring program.31,32 Prevention is the best cure, and many critical care units have adopted a “bundle” approach to eliminate CAUTI.33,34 The key components of CAUTI prevention are listed below26: 1. Avoid unnecessary use of urinary catheters 2. Insert urinary catheters using aseptic technique 3. Adopt evidence-based standards for maintenance of urinary catheters 4. Review the need for the urinary catheter daily and remove promptly Interventions to prevent CAUTI are listed in the Box 27-3. Patient Safety Alert Prevention of Catheter-Associated Urinary Tract infections (CAUTI) 2 Inset Urinary Catheters Using Aseptic Technique Sterile Technique and Sterile Equipment Use standard supply kits that contain all necessary items: • Sterile gloves, drape, sponges, antiseptic solution for cleaning the meatus, single-use packet of lubricant jelly for insertion • Use as small a catheter as possible to minimize urethral trauma • Single attempt to insert urinary catheter • Use new catheter if second attempt at catheterization is required 3 Adopt Evidence-Based Standards for Maintenance of Urinary Catheters Maintenance of a Closed Drainage System Maintain a sterile closed drainage system Maintain unobstructed urine flow (avoid dependent loops) Keep drainage bag below the level of the bladder at all times Do not allow drainage bag to touch the floor Empty the collection bag regularly, using a separate container for each patient Do not allow the drainage spigot to touch the collection container Do not break the system to collect a urine sample. Collect from sampling port in the tubing drainage system, disinfecting the port and aspirating using aseptic technique Avoid catheter irrigation except in the case of an obstructed catheter; use a bladder ultrasound scan to determine if there is urine in the bladder Routine scheduled replacement of catheters is not recommended 4 Review the Need for the Urinary Catheter Daily and Remove Promptly Hospital Strategies to Ensure Early Removal of Urinary Catheters Based on data from Gould CV, et al. Guideline for prevention of catheter associated urinary tract infections 2009. Centers for Disease Control and Prevention. http://www.cdc.gov/hicpac/pdf/cauti/cautiguideline2009final.pdf. 2009. Accessed September 15, 2012. 1. Fluid retention caused by inadequate urine output. 2. Low serum albumin levels create a lower oncotic pressure in the vasculature, and more fluid seeps out into the interstitial spaces to cause peripheral edema. 3. Inflammation associated with AKI or a coexisting nonrenal disease increases vascular permeability, facilitating fluid movement from the vessels into interstitial spaces. Electrolyte levels require frequent observation, especially in the critical phases of AKI when potassium can quickly reach levels of 6.0 mEq/L or higher (see Table 27-4). Specific electrocardiographic changes are associated with hyperkalemia: peaked T waves, a widening of the QRS interval, and ultimately, ventricular tachycardia or fibrillation.36 If hyperkalemia is identified, all potassium supplements are stopped. If the patient is producing urine, IV diuretics can be administered. Acute hyperkalemia can be treated temporarily by IV administration of insulin and glucose. An infusion of 50 mL of 50% dextrose accompanied by 10 units of regular insulin forces potassium out of the serum and into the cells. To treat smaller increases in serum potassium, nonabsorbable potassium-binding resins may be used.37 The binding resins can be administered orally, through a nasogastric tube, or rectally, to treat hyperkalemia. Cation exchange resins employ either sodium (Kayexalate) or calcium (Sorbisterit, Ca-Resonium, Argamate) and exchange the cation for potassium across the gastrointestinal wall.38 The potassium is contained in the lower gastrointestinal tract and is eliminated with the stool. Potassium-binding resins and dialysis are the only permanent methods of potassium removal to treat hyperkalemia. Alterations in sodium level are an expected finding in kidney failure (see Table 27-4). Both hypernatremia (elevated serum sodium) and hyponatremia (low serum sodium) are associated with increased mortality with kidney failure39; this is unrelated to whether the patient has a diagnosis of heart failure or not.39
Kidney Disorders and Therapeutic Management
Acute Kidney Injury
Critical Illness and Acute Kidney Injury
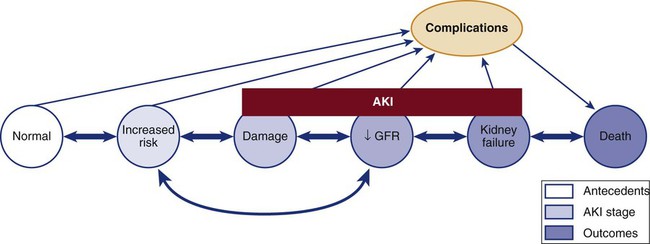
Definitions of Acute Kidney Injury
RIFLE Criteria
RIFLE
SERUM CREATININE CRITERIA*
URINE OUTPUT CRITERIA
Risk
Serum Cr increased 1.5 times above normal or
UO <0.5 mL/kg/hr for 6 hr
Serum Cr increase ≥0.3 mg/dL
Injury
Serum Cr increased 2 times above normal
UO <0.5 mL/kg/hr for 12 hr
Failure
Serum Cr increased 3 times above normal or
UO <0.3 mL/kg/hr for 24 hr or anuria for 12 hr (oliguria)
Serum Cr ≥4 mg/dL or
Serum Cr acute rise ≥0.5 mg/dL
Loss
Persistent AKI = complete loss of kidney function for more than 4 wk
ESKD
End-stage kidney disease
Acute Kidney Injury Network Criteria
Types of Acute Kidney Injury
Prerenal Acute Kidney Injury
Intrarenal Acute Kidney Injury
Postrenal Acute Kidney Injury
Assessment and Diagnosis
Laboratory Assessment
ELECTROLYTE
NORMAL VALUE
Sodium
135-145 mEq/L
Potassium
3.5-4.5 mEq/L
Chloride
98-108 mEq/L
Calcium
8.5-10.5 mg/dL or 4.5-5.8 mEq/L
Phosphorus
2.7-4.5 mg/dL
Magnesium
1.5-2.5 mEq/L
Bicarbonate
24-28 mEq/L
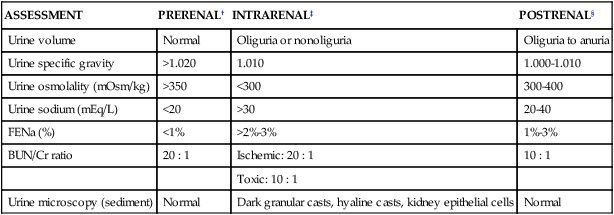
ASSESSMENT
PRERENAL†
INTRARENAL‡
POSTRENAL§
Urine volume
Normal
Oliguria or nonoliguria
Oliguria to anuria
Urine specific gravity
>1.020
1.010
1.000-1.010
Urine osmolality (mOsm/kg)
>350
<300
300-400
Urine sodium (mEq/L)
<20
>30
20-40
FENa (%)
<1%
>2%-3%
1%-3%
BUN/Cr ratio
20 : 1
Ischemic: 20 : 1
10 : 1
Toxic: 10 : 1
Urine microscopy (sediment)
Normal
Dark granular casts, hyaline casts, kidney epithelial cells
Normal
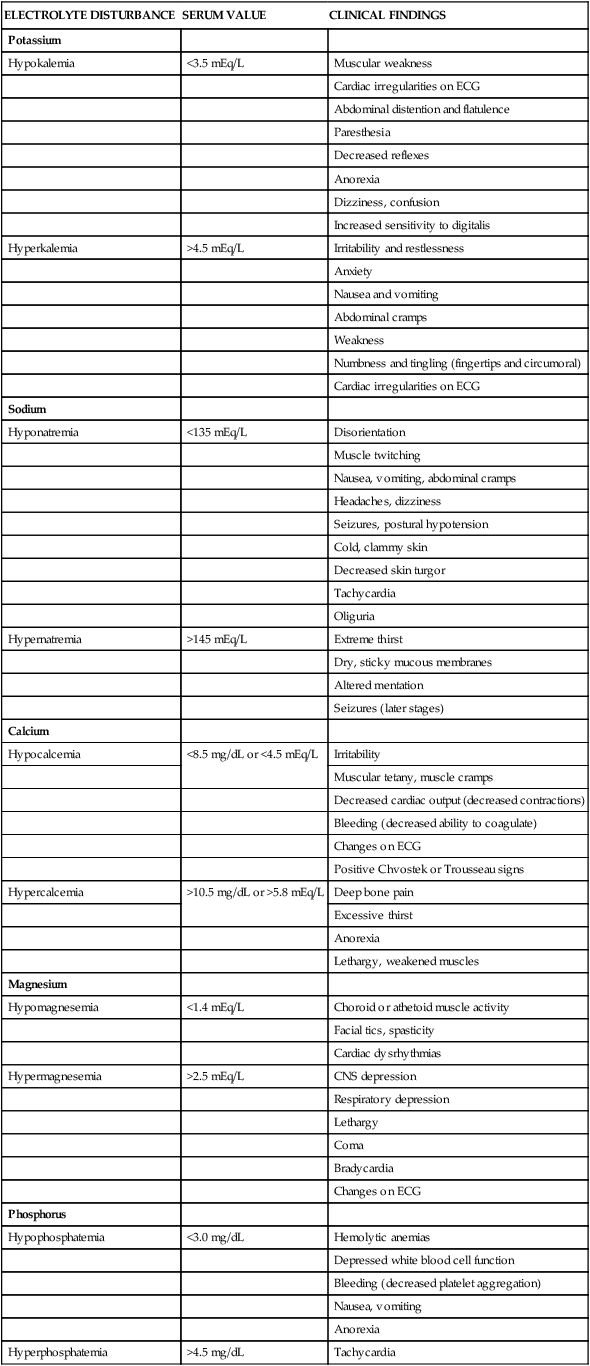
ELECTROLYTE DISTURBANCE
SERUM VALUE
CLINICAL FINDINGS
Potassium
Hypokalemia
<3.5 mEq/L
Muscular weakness
Cardiac irregularities on ECG
Abdominal distention and flatulence
Paresthesia
Decreased reflexes
Anorexia
Dizziness, confusion
Increased sensitivity to digitalis
Hyperkalemia
>4.5 mEq/L
Irritability and restlessness
Anxiety
Nausea and vomiting
Abdominal cramps
Weakness
Numbness and tingling (fingertips and circumoral)
Cardiac irregularities on ECG
Sodium
Hyponatremia
<135 mEq/L
Disorientation
Muscle twitching
Nausea, vomiting, abdominal cramps
Headaches, dizziness
Seizures, postural hypotension
Cold, clammy skin
Decreased skin turgor
Tachycardia
Oliguria
Hypernatremia
>145 mEq/L
Extreme thirst
Dry, sticky mucous membranes
Altered mentation
Seizures (later stages)
Calcium
Hypocalcemia
<8.5 mg/dL or <4.5 mEq/L
Irritability
Muscular tetany, muscle cramps
Decreased cardiac output (decreased contractions)
Bleeding (decreased ability to coagulate)
Changes on ECG
Positive Chvostek or Trousseau signs
Hypercalcemia
>10.5 mg/dL or >5.8 mEq/L
Deep bone pain
Excessive thirst
Anorexia
Lethargy, weakened muscles
Magnesium
Hypomagnesemia
<1.4 mEq/L
Choroid or athetoid muscle activity
Facial tics, spasticity
Cardiac dysrhythmias
Hypermagnesemia
>2.5 mEq/L
CNS depression
Respiratory depression
Lethargy
Coma
Bradycardia
Changes on ECG
Phosphorus
Hypophosphatemia
<3.0 mg/dL
Hemolytic anemias
Depressed white blood cell function
Bleeding (decreased platelet aggregation)
Nausea, vomiting
Anorexia
Hyperphosphatemia
>4.5 mg/dL
Tachycardia
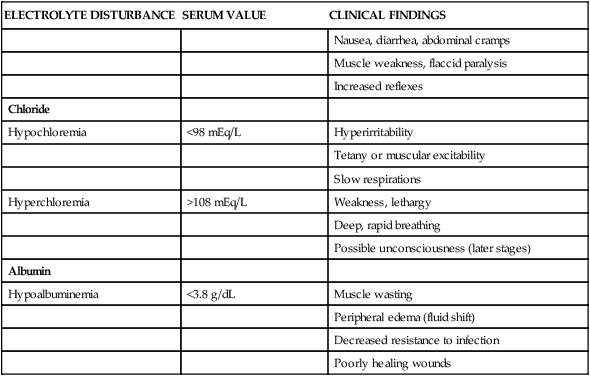
Nausea, diarrhea, abdominal cramps
Muscle weakness, flaccid paralysis
Increased reflexes
Chloride
Hypochloremia
<98 mEq/L
Hyperirritability
Tetany or muscular excitability
Slow respirations
Hyperchloremia
>108 mEq/L
Weakness, lethargy
Deep, rapid breathing
Possible unconsciousness (later stages)
Albumin
Hypoalbuminemia
<3.8 g/dL
Muscle wasting
Peripheral edema (fluid shift)
Decreased resistance to infection
Poorly healing wounds
Acidosis
Blood Urea Nitrogen
Serum Creatinine
Creatinine Clearance
Fractional Excretion of Sodium
At-Risk Disease States and Acute Kidney Injury
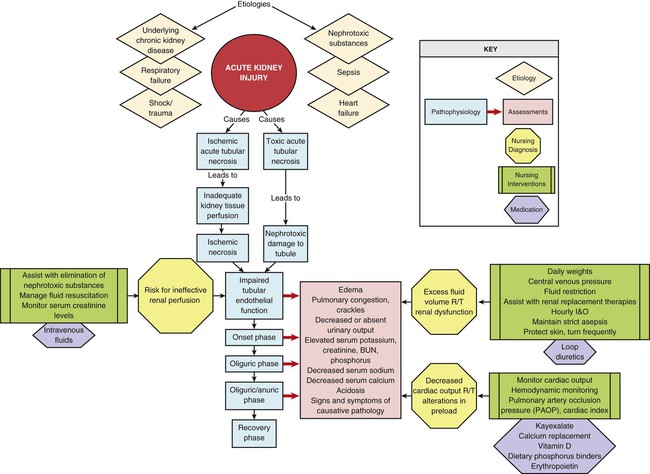
Underlying Chronic Kidney Disease
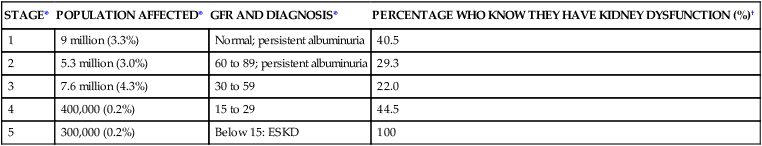
STAGE*
POPULATION AFFECTED*
GFR AND DIAGNOSIS*
PERCENTAGE WHO KNOW THEY HAVE KIDNEY DYSFUNCTION (%)†
1
9 million (3.3%)
Normal; persistent albuminuria
40.5
2
5.3 million (3.0%)
60 to 89; persistent albuminuria
29.3
3
7.6 million (4.3%)
30 to 59
22.0
4
400,000 (0.2%)
15 to 29
44.5
5
300,000 (0.2%)
Below 15: ESKD
100
Heart Failure and Acute Kidney Injury
Respiratory Failure and Acute Kidney Injury
Sepsis and Acute Kidney Injury
Trauma and Acute Kidney Injury
Trauma Admissions
Rhabdomyolysis
Contrast-Induced Nephrotoxic Injury and Acute Kidney Injury
Promote Hydration and Avoid Dehydration
Medications
Catheter-Associated Urinary Tract Infection
 Box 27-3
Box 27-3
Hemodynamic Monitoring and Fluid Balance
Physical Assessment
Electrolyte Balance
Potassium
Sodium
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Kidney Disorders and Therapeutic Management
 Avoid Unnecessary Use of Indwelling Urinary Catheters
Avoid Unnecessary Use of Indwelling Urinary Catheters
Get Clinical Tree app for offline access
