(EYE-bue-PROE-fen)
Caldolor
NSAID
pH 7.4
Usual dose
Use the lowest effective dose for the shortest duration of time based on individual needs and response. Re-evaluate after the initial dose. Adjust dose and frequency as indicated. Total daily dose should not exceed 3,200 mg. Adequate hydration and correction of hypovolemia required before administration to reduce the risk of adverse renal reactions.
Analgesia:
400 to 800 mg every 6 hours as necessary.
Antipyretic:
400 mg every 4 to 6 hours or 100 to 200 mg every 4 hours as necessary.
Pediatric dose
| Pediatric Dosing of Ibuprofen for Fever and Pain | |||
| Age-Group | Dose | Dosing Interval | Maximum Daily Dose* |
| 6 months to less than 12 years | 10 mg/kg up to 400 mg max | Every 4 to 6 hr as needed | 40 mg/kg or 2,400 mg |
| 12 to 17 years | 400 mg | Every 4 to 6 hr as needed | 2,400 mg |

*Maximum daily dose is 40 mg/kg or 2,400 mg, whichever is less.
See comments under Usual Dose and Maternal/Child.
Dose adjustments
To minimize the potential risk for an adverse cardiovascular (CV) event, use the lowest effective dose for the shortest duration possible. ■ Lower-end initial and reduced doses may be indicated in the elderly and/or debilitated. Consider potential for decreased organ function and concomitant disease or drug therapy.
Dilution
Must be diluted to a final concentration of 4 mg/mL or less for both adult and weight-based pediatric dosing. Infusion without dilution can cause hemolysis. Further dilute in NS, D5W, or LR.
Filters:
Specific information not available; consult pharmacist.
Storage:
Store vials at CRT. Diluted solutions stable for up to 24 hours at 20° to 25° C (68° to 77° F) and with ambient room lighting.
Compatibility
Specific information not available; consult pharmacist.
Rate of administration
Adult:
A single dose as an infusion over no less than 30 minutes.
Pediatric:
A single dose as an infusion over no less than 10 minutes.
Actions
A nonsteroidal anti-inflammatory drug (NSAID) that has anti-inflammatory, analgesic, and antipyretic activity. Mechanism of action is not completely understood but involves inhibition of cyclooxygenase (COX-1 and COX-2). Ibuprofen is a mixture of two isomers: [-]R- and [+]S-. The [+]S- isomer is responsible for the activity of ibuprofen. The [-]R- isomer slowly converts to the active [+]S- isomer to maintain levels of the active drug in the circulation. Highly protein bound; the mean half-life of a 400-mg dose is 2.2 hours, and the mean half-life of an 800-mg dose is 2.44 hours. Metabolized in the liver via oxidation and excreted in the urine.
Indications and uses
Management of mild to moderate pain. ■ Management of moderate to severe pain as an adjunct to opioid analgesics. ■ Reduction of fever.
Contraindications
Known hypersensitivity (anaphylactoid reactions, serious skin reactions) to ibuprofen or other NSAIDs. ■ Known history of asthma, urticaria, or allergic-type reactions after taking aspirin or other NSAIDs. ■ In the setting of coronary artery bypass graft (CABG) surgery. ■ See Maternal/Child.
Precautions
For IV infusion only. ■ NSAIDs increase the risk for serious cardiovascular (CV) thrombotic events, including MI and stroke, which can be fatal. This risk may occur early in treatment and may increase with duration of use. The increase in CV thrombotic risk has been observed most consistently at higher doses. ■ Avoid the use of ibuprofen in patients with a recent MI or in patients with heart failure unless the benefits are expected to outweigh the risks. Studies have shown these patients to be at increased risk for reinfarction, CV-related death, and all-cause mortality. ■ NSAIDs increase the risk for serious GI ulceration, bleeding, and/or perforation. Can occur at any time, with or without warning symptoms, and can be fatal. Use extreme caution in patients with a prior history of peptic ulcer disease or GI bleeding. Risk is also increased in elderly or debilitated patients, in patients with advanced liver disease, and with concomitant use of aspirin, oral corticosteroids, anticoagulants, alcohol, selective serotonin reuptake inhibitors (SSRIs), or smoking. ■ May cause elevations of liver function tests (e.g., ALT, AST). Rare cases of serious, sometimes fatal, hepatic injury (fulminant hepatitis, liver necrosis, hepatic failure) have occurred. ■ May cause fluid retention and edema; use caution in patients with CHF or edema; see Drug/Lab Interactions. ■ May precipitate hypertension or worsen pre-existing hypertension; see Drug/Lab Interactions. ■ In addition to the usual caution in patients with reduced hepatic or renal function, NSAIDs may cause a dose-dependent reduction in renal prostaglandin formation and, secondarily, in renal blood flow, which can precipitate renal failure. Patients with impaired renal function, heart failure, liver dysfunction, dehydration, or hypovolemia; elderly patients; and patients receiving ACE inhibitors (e.g., enalapril [Vasotec], lisinopril [Zestril]), angiotensin receptor blockers (ARBs) (e.g., losartan [Cozaar], valsartan [Diovan]), or diuretics are at greatest risk. ■ Avoid ibuprofen use in patients with advanced renal disease unless the benefits are expected to outweigh the risk. ■ Increases in serum potassium, including hyperkalemia, have been reported. ■ Anaphylactic reactions have occurred in patients with and without known hypersensitivity to ibuprofen and in patients with aspirin-sensitive asthma. Cross-reactivity between aspirin and other NSAIDs has been reported in aspirin-sensitive patients; see Contraindications. Use caution in patients with pre-existing asthma (without known aspirin sensitivity), and monitor for changes in S/S of asthma. ■ May cause serious skin reactions (e.g., exfoliative dermatitis, Stevens-Johnson syndrome, toxic epidermal necrolysis) without warning; some have been fatal. ■ Anti-inflammatory and antipyretic effects may reduce the utility of these diagnostic signs in detecting infections. ■ May cause anemia. Anemia may be due to occult or gross GI blood loss, fluid retention, or an incompletely described effect on erythropoiesis. ■ NSAIDs inhibit platelet aggregation and may increase the risk of bleeding; see Drug/Lab Interactions. ■ Aseptic meningitis and ophthalmologic effects (e.g., blurred or diminished vision, changes in color vision) have been reported with oral ibuprofen. ■ See Maternal/Child.
Monitor:
Correct hypovolemia before administration and maintain adequate hydration. ■ Obtain baseline vital signs and monitor frequently during therapy. CBC, electrolytes, liver function tests, and SCr or CrCl may be indicated for a baseline or as needed as symptoms develop. ■ Monitor patients with or without a previous history of CV disease for S/S of CV events (e.g., chest pain, dyspnea, edema, hypertension, limb or facial paralysis). ■ Monitor for edema or signs of worsening heart failure. ■ Observe for S/S of GI ulceration or bleeding and/or liver dysfunction. ■ Monitor renal function, especially in patients with impaired renal function. ■ Monitor for S/S of hypersensitivity reactions (e.g., anaphylaxis, pruritus, rash, urticaria, or wheezing). ■ Monitor patients who may be adversely affected by alterations in platelet function (e.g., patients with coagulation disorders or patients receiving anticoagulants).
Patient education:
Side effects have resulted in extended hospitalization and could be fatal. ■ Promptly report any S/S of CV thrombotic events (e.g., chest pain, shortness of breath, slurred speech, weakness), GI adverse events (e.g., dyspepsia, epigastric pain, hematemesis, melena), hepatotoxicity (e.g., diarrhea, fatigue, flu-like symptoms, jaundice, lethargy, nausea, right upper quadrant tenderness), heart failure (e.g., edema, shortness of breath, weight gain), and/or a hypersensitivity reaction (e.g., difficulty breathing, swelling of the face or throat) or any type or rash. ■ Avoid use of concomitant NSAIDs (e.g., naproxen [Aleve, Naprosyn], celecoxib [Celebrex]). Over-the-counter medications for the treatment of fever, cold, and insomnia may contain NSAIDs. Consult pharmacist before use. ■ Discuss use of low-dose aspirin for cardiac prophylaxis with health care provider before initiating concomitant therapy; see Maternal/Child.
Maternal/child:
Avoid use starting at 30 weeks’ gestation; premature closure of the ductus arteriosus in the fetus may occur. Use before 30 weeks’ gestation only if the potential benefit justifies the potential risk to the fetus. ■ Effects during labor and delivery unknown. ■ Use with caution during breast-feeding. ■ Safety and effectiveness for use in pediatric patients under 6 months of age not established.
Elderly:
Increased risk for serious NSAID-associated GI, cardiovascular, and/or renal adverse events. ■ Dosing should be cautious; see Dose Adjustments.
Drug/lab interactions
Concurrent use with aspirin or other salicylates (e.g., diflunisal, salsalate) is not recommended; may increase the risk of toxicity and serious GI events. ■ Synergistic effect on bleeding with anticoagulants (e.g., heparin, warfarin [Coumadin]), antiplatelet agents (e.g., aspirin), SSRIs (e.g., fluoxetine [Prozac], paroxetine [Paxil], sertraline [Zoloft]), and serotonin-norepinephrine reuptake inhibitors (SNRIs) (e.g., duloxetine [Cymbalta], venlafaxine [Effexor]); risk of bleeding increased. Monitor patients closely if concomitant use indicated. ■ NSAIDs may decrease the effectiveness of ACE inhibitors (e.g., enalapril [Vasotec], lisinopril [Zestril]), angiotensin receptor blockers (ARBs) (e.g., losartan [Cozaar], valsartan [Diovan]), and beta-blockers (e.g., metoprolol [Lopressor], propranolol [Inderal]); monitor BP. ■ Coadministration with an ACE inhibitor or ARB may result in deterioration of renal function in at-risk patients; monitor renal function. ■ Ibuprofen can reduce the natriuretic effects of loop diuretics (e.g., furosemide [Lasix]) and thiazide diuretics (e.g., hydrochlorothiazide); observe for signs of worsening renal function and ensure diuretic/therapeutic effectiveness. ■ May increase digoxin serum concentration and prolong half-life of digoxin; monitor digoxin levels with concomitant use. ■ Concurrent use of lithium with NSAIDs may decrease lithium clearance, increasing plasma levels of lithium; observe for signs of lithium toxicity. ■ Concurrent use of NSAIDs with methotrexate may enhance methotrexate toxicity (e.g., neutropenia, thrombocytopenia, renal dysfunction); monitor for signs of methotrexate toxicity. ■ Concomitant use with cyclosporine (Sandimmune) may increase cyclosporine’s nephrotoxicity; monitor renal function. ■ Concomitant use with pemetrexed (Alimta) may increase the risk of pemetrexed-associated myelosuppression and renal and GI toxicity; avoid concomitant use for 2 to 5 days before pemetrexed administration and for 2 days following administration in patients with mild to moderate renal impairment (CrCl 45 to 79 mL/min). See pemetrexed monograph.
Side effects
Adults:
The most common side effects are dizziness, flatulence, headache, hemorrhage, and nausea and vomiting. Other side effects include abdominal discomfort, anemia, bacteremia, bacterial pneumonia, cough, diarrhea, dyspepsia, eosinophilia, hyperkalemia, hypernatremia, hypertension, hypoalbuminemia, hypokalemia, hypoproteinemia, hypotension, increased blood urea, increased lactic dehydrogenase (LDH), neutropenia, peripheral edema, thrombocytosis, urinary retention, wound hemorrhage. See Precautions for potential major side effects.
Pediatric patients:
The most common side effects are anemia, headache, infusion site pain, nausea, and vomiting.
Overdose:
Acute renal failure, coma, drowsiness, epigastric pain, GI bleeding, hypertension, lethargy, nausea, respiratory depression, and vomiting.
Antidote
Keep the physician informed of significant side effects. With increasing severity or onset of symptoms of any major side effect (e.g., CHF; edema; hypersensitivity reactions; GI bleeding, ulceration, or perforation; hepatic or renal effects; hypertension; skin reactions; thrombotic events), discontinue the drug and notify the physician. A patent airway, artificial ventilation, oxygen therapy, and other symptomatic treatment must be instituted promptly if indicated. Treat anaphylaxis with epinephrine (Adrenalin), diphenhydramine (Benadryl), and corticosteroids as indicated. No known antidote. Forced diuresis, alkalinization of urine, hemodialysis, or hemoperfusion may not be useful due to high protein binding.
Ibuprofen lysine
(eye-byou-PROH-fen LIE-seen)
NeoProfen
NSAID (patent ductus arteriosus adjunct)
pH 7
Neonatal dose
All doses are based on birth weight. A course of therapy is three doses given at 24-hour intervals.
Initial dose:
10 mg/kg. Follow with a dose of 5 mg/kg in 24 hours and repeat at 48 hours; see Dose Adjustments.
After completion of the first course, no further doses are indicated if the ductus arteriosus closes or is significantly reduced in size. If the ductus arteriosus fails to close or reopens, a second course, alternative pharmacologic therapy, or surgery may be necessary.
Dose adjustments
If urine output is less than 0.6 mL/kg/hr at any time a dose is to be given, withhold dose until lab studies confirm a return to normal renal function.
Dilution
Each single dose must be diluted to an appropriate volume for administration as an infusion over 15 minutes with dextrose or saline. Prepare for infusion and begin administration within 30 minutes of preparation. A fresh solution should be prepared just before each administration. Contains no preservatives; discard any unused portion.
Filters:
Specific information not available.
Storage:
Store vials in cartons at CRT and protect from light until use. Use reconstituted solution within 30 minutes of preparation.
Compatibility
Manufacturer states, “Should not be simultaneously administered in the same IV line with Total Parenteral Nutrition” (TPN). If required, interrupt TPN for 15 minutes before and after ibuprofen administration. Maintain IV line patency with dextrose or saline infusion.
Rate of administration
Administer via the IV port nearest the insertion site.
A single dose, properly diluted, and infused continuously over 15 minutes.
Actions
A nonsteroidal anti-inflammatory drug (NSAID). Mechanism of action by which it causes closure of a patent ductus arteriosus (PDA) is unknown; however, in adults it is an inhibitor of prostaglandin synthesis. By closing the patent ductus arteriosus, the need for surgical intervention is eliminated. Half-life varies inversely with postnatal age, but in general the half-life in infants is more than 10 times longer than in adults. In lower-birth-weight premature infants, half-life may range from 20 to 51 hours. Metabolism and excretion have not been studied in premature infants. In adults, it is metabolized in the liver and excreted in the urine and feces.
Indications and uses
Closure of a clinically significant patent ductus arteriosus in premature infants weighing between 500 and 1,500 Gm who are no more than 32 weeks’ gestational age when usual medical management (e.g., fluid restriction, diuretics, respiratory support) is not effective. Consequences beyond 8 weeks after treatment have not been evaluated.
Contraindications
Bleeding (especially active intracranial hemorrhage or GI bleeding), coagulation defects, suspected necrotizing enterocolitis, infants with congenital heart disease (e.g., pulmonary atresia, severe coarctation of the aorta, severe tetralogy of Fallot) who require patency of the ductus arteriosus for satisfactory pulmonary or systemic blood flow, proven or suspected untreated infection, significant renal impairment, thrombocytopenia.
Precautions
For IV use only; see Monitor. ■ Reserve for infants with clear evidence of a clinically significant PDA. ■ For use only in a highly supervised setting such as an intensive care nursery. ■ No long-term evaluations available. Effects of ibuprofen on neurodevelopmental outcome and growth and on disease processes associated with prematurity (e.g., retinopathy of prematurity, chronic lung disease) have not been assessed. ■ May alter the usual signs of infection. Use with extreme caution in the presence of an existing controlled infection and in infants at risk for infection. ■ Can inhibit platelet aggregation. ■ Can prolong bleeding time in adults; use caution in infants with underlying hemostatic defects; see Contraindications. ■ Can displace bilirubin from albumin-binding sites; use with caution in infants with elevated total bilirubin.
Monitor:
Confirm absolute patency of vein. Avoid extravasation; will irritate tissue. Administration via an umbilical arterial line has not been studied. ■ Obtain baseline and monitor vital signs, oxygenation, acid-base status, fluid and electrolyte balance, and kidney function (SCr, BUN, urine output). ■ Can cause a reduction in urine output, increased BUN and SCr, and a decreased CrCl; may progress to oliguria or renal failure. Monitor all infants closely, especially those with some degree of renal impairment. ■ May inhibit platelet aggregation; monitor for signs of bleeding. ■ Monitor for signs of infection.
Drug/lab interactions
Drug interactions have not been studied.
Side effects
The most commonly reported side effects include adrenal insufficiency, anemia, apnea, atelectasis, decreased urine output, edema, GI disorders (including nonnecrotizing enterocolitis), hematuria, hypernatremia, hypocalcemia, hypoglycemia, increased BUN and SCr, intraventricular hemorrhage and other bleeding, renal failure, renal insufficiency, respiratory failure, respiratory infection, sepsis, skin lesion or irritation, and urinary tract infection. Other side effects of unknown association include abdominal distension, cardiac failure, cholestasis, convulsions, feeding problems, gastritis, gastroesophageal reflux, hypotension, ileus, infections, inguinal hernia, injection site reactions, jaundice, lab abnormalities (e.g., hyperglycemia, neutropenia, thrombocytopenia), and tachycardia.
Post-marketing:
GI perforation, necrotizing enterocolitis, and pulmonary hypertension.
Overdose:
Breathing difficulties, coma, drowsiness, hypotension, irregular heartbeat, kidney failure, seizures, and vomiting have occurred in individuals (not necessarily in premature infants) following overdose of oral ibuprofen.
Antidote
Discontinue the drug and notify the physician of all side effects. Based on severity, side effects may be treated symptomatically or drug will be completely discontinued in favor of surgical intervention. In case of overdose, there is no specific antidote. Treat symptomatically and follow for several days after apparent recovery; GI ulceration and hemorrhage may occur. Resuscitate as necessary.
Ibutilide fumarate 
(ih-BYOU-tih-lyd FU-mar-ayt)
Corvert
Antiarrhythmic
pH 4.6
Usual dose
| Guidelines for Ibutilide Dosing | ||
| Patient Weight | Initial Infusion (over 10 minutes) | Second Infusion |
| 60 kg (132 lb) or more | 1 mg ibutilide fumarate (one vial [10 mL]) | If the arrhythmia does not terminate within 10 minutes after the end of the initial infusion, a second 10-minute infusion of equal strength may be administered 10 minutes after completion of the first infusion. |
| Less than 60 kg (132 lb) | 0.01 mg/kg ibutilide fumarate (0.1 mL/kg) | |

Discontinue infusion promptly when the presenting arrhythmia is terminated (desired effect). Must also be discontinued immediately if sustained or nonsustained ventricular tachycardia or marked prolongation of QT or QTc occurs (adverse effects). Postconversion treatment with appropriate antiarrhythmics (e.g., digoxin, verapamil, or propranolol) is usually required.
Dose adjustments
Dose selection should be cautious in the elderly. Reduced doses may be indicated based on the potential for decreased organ function and concomitant disease or drug therapy. ■ No adjustments required in patients with impaired hepatic or renal function; see Monitor. Lower doses may be indicated in post–cardiac surgery patients. In recent studies one or two infusions of 0.5 mg in patients weighing 60 kg or more or 0.005 mg/kg/dose for patients under 60 kg was effective in terminating atrial fibrillation and/or flutter.
Dilution
May be given undiluted or may be diluted in 50 mL of NS or D5W and given as an infusion. 1 mg (10 mL of a 0.1-mg/mL solution) of ibutilide in 50 mL diluent yields 0.017 mg/mL.
Storage:
Store at CRT in carton until use. Stable after dilution for 24 hours at room temperature, 48 hours if refrigerated.
Compatibility
Manufacturer lists as compatible with NS and D5W packaged in glass, polyvinyl chloride, or polyolefin infusion containers. Additional information not available; consult pharmacist.
Rate of administration
A single dose by injection or infusion over 10 minutes.
Actions
A Class III antiarrhythmic agent that produces mild slowing of the sinus rate and atrioventricular conduction. Delays repolarization by activation of a slow inward current (sodium) rather than blocking outward potassium currents. Prolonged atrial and ventricular action potential duration and refractoriness result. Produces dose-related prolongation of the QT interval (may result from dose of ibutilide or rate of injection). Conversion of atrial flutter/fibrillation usually occurs within 30 minutes but may take up to 90 minutes after the start of the infusion. Most patients remain in normal sinus rhythm (NSR) for 24 hours. At recommended doses, ibutilide has no clinically significant effects on cardiac output, mean pulmonary arterial pressure, or pulmonary capillary wedge pressure. Rapidly distributed and metabolized. Elimination half-life is 6 hours (range 2 to 12 hours). Primarily excreted in urine (7% as unchanged drug). Excreted in small amounts in feces.
Indications and uses
Rapid conversion of recent-onset atrial fibrillation or atrial flutter to sinus rhythm. Patients with more recent onset of arrhythmia have a higher rate of conversion. Effectiveness was less in those with a longer-duration arrhythmia.
Contraindications
Known hypersensitivity to ibutilide or any of its components. ■ Not recommended in patients who have had a previous polymorphic ventricular tachycardia (e.g., torsades de pointes). ■ See Drug/Lab Interactions.
Precautions
For IV infusion only. ■ Usually administered by or under the direction of the physician specialist. ■ Skilled personnel and proper equipment (e.g., cardiac monitors, intracardiac pacing facilities, cardioverter/defibrillator, emergency drugs) must be immediately available. ■ May cause life-threatening arrhythmias (e.g., torsades de pointes) with or without documented QT prolongation. ■ Correct hypokalemia and hypomagnesemia before use; may exaggerate a prolonged QT and cause arrhythmias. ■ Adequate anticoagulation (usually at least 2 weeks) is required for any patient with atrial fibrillation of more than 2 to 3 days’ duration. ■ Select patients carefully; benefits (potential for maintaining sinus rhythm) must outweigh risks. Patients with chronic atrial fibrillation are more likely to revert back to atrial fibrillation after conversion to sinus rhythm. Patients with a QTc interval greater than 440 msec or a serum potassium less than 4.0 mEq/L are at very high risk to develop life-threatening arrhythmias. ■ Patients with a history of CHF may be more susceptible to sustained polymorphic VT. ■ Slightly more effective in atrial flutter than atrial fibrillation. ■ See Drug/Lab Interactions.
Monitor:
Obtain weight, baseline vital signs, and ECG before administration. Continuous ECG monitoring during and after infusion indicated to observe for arrhythmias. Watch for QT or QTc prolongation; may cause arrhythmia (torsades de pointes) with or without QT prolongation. ■ Monitor BP and HR. Bradycardia, a varying HR, and/or hypokalemia may increase risk of arrhythmia. ■ Arrhythmia occurs most frequently within 40 minutes of completion of infusion but may occur for up to 3 hours after infusion. Monitor ECG for a minimum of 4 hours or until QTc has returned to baseline. Monitor longer if there are any episodes of arrhythmias or if the patient has impaired liver function.
Patient education:
Report promptly any feeling of faintness, difficulty breathing, or pain or stinging along injection site.
Maternal/child:
Category C: benefits must outweigh risks. Caused birth defects and was embryocidal in rats. ■ Temporarily discontinue breast-feeding. ■ Safety and effectiveness for use in pediatric patients under 18 years of age not established.
Elderly:
No age-related differences observed. Median age in clinical trials was 65 years. ■ Lower-end initial doses may be indicated; see Dose Adjustments.
Drug/lab interactions
Should not be given concurrently with Class Ia antiarrhythmics (e.g., disopyramide [Norpace], procainamide [Pronestyl], quinidine) or other Class III antiarrhythmics (e.g., amiodarone [Nexterone], sotalol [Betapace]). Withhold any of these agents for at least 5 half-lives before ibutilide infusion and for 4 hours after. ■ Incidence of arrhythmia may be increased with other drugs that prolong the QT interval (e.g., phenothiazines [e.g., promethazine (Phenergan)], tricyclic antidepressants [e.g., amitriptyline (Elavil)], tetracyclic antidepressants [e.g., maprotiline (Ludiomil)]). ■ Monitor serum digoxin levels to avoid digoxin toxicity. ■ Use with digoxin, beta-blockers, or calcium channel blockers does not alter safety or effectiveness of ibutilide. However, sotalol is a beta-blocker, and its use with ibutilide is restricted because it has Class III antiarrhythmic activity; see first sentence above.
Side effects
Sustained polymorphic VT (1.7%) and nonsustained polymorphic VT (2.7%) can deteriorate into ventricular fibrillation and be fatal. May cause many other arrhythmias (e.g., first-, second-, or third-degree AV block [1.5%], bradycardia [1.2%], bundle branch block [1.9%], nonsustained monomorphic VT [4.9%], prolonged QT segment [1.2%], PVCs [5.1%], tachycardia [2.7%]), CHF (0.5%), headache (3.6%), hypertension (1.2%), hypotension (2%), nausea (1.9%), palpitation (1%).
Overdose:
Side effects exaggerated with overdose in humans. Acute overdose in animals resulted in CNS depression, rapid gasping breathing, convulsions.
Antidote
If proarrhythmias occur, discontinue ibutilide; correct electrolyte abnormalities (e.g., potassium and magnesium). Overdrive cardiac pacing, electrical cardioversion, or defibrillation may be required. Infusions of magnesium sulfate may be helpful. Avoid treatment with antiarrhythmic agents. VT that deteriorates to VF will require immediate defibrillation.
Idarubicin hydrochloride 
(eye-dah-ROOB-ih-sin hy-droh-KLOR-eyed)
Idamycin PFS, Idarubicin PFS, IDR
Antineoplastic (anthracycline antibiotic)
pH 5 to 7
Usual dose
Adult acute myeloid leukemia (AML) induction therapy:
Induction:
Idarubicin 12 mg/M2/day for 3 days. Used in combination with cytarabine (Ara-C). Cytarabine 100 mg/M2/day may be given as a continuous infusion on Days 1 to 7 (daily for 7 days), or alternately the cytarabine may be given as an IV injection of 25 mg/M2 followed immediately by a continuous infusion of cytarabine 200 mg/M2/day on Days 1 to 5 (daily for 5 days). See Precautions/Monitor. If unequivocal evidence of leukemia remains after the first course, a second course may be given; see Dose Adjustments. The benefit of an aggressive consolidation and maintenance program in prolonging the duration of remissions and survival has not been proven. See prescribing information for regimens used in clinical trials.
Dose adjustments
Delay second course until full recovery if severe mucositis has occurred and reduce dose by 25%. ■ Consider dose reductions in impaired liver and kidney function based on bilirubin and/or creatinine levels above the normal range. Do not administer if bilirubin is above 5 mg/dL. In one Phase III clinical trial, patients with bilirubin levels between 2.6 and 5 mg/dL received a 50% reduction in dose.
Dilution
Specific techniques required (see precautions).
Idamycin PFS is a liquid formulation; each 5 mg of powdered idamycin must be reconstituted with 5 mL of nonbacteriostatic NS for injection (1 mg/mL). Use extreme caution inserting the needle; vial contents are under negative pressure. Avoid any possibility of inhalation from aerosol or any skin contamination.
Filters:
No data available from manufacturer.
Storage:
PFS product must be refrigerated. Reconstituted idamycin is stable for 7 days under refrigeration (2° to 8° C [36° to 46° F]) or 3 days (72 hours) at room temperature (15° to 30° C [59° to 86° F]). Discard unused solution appropriately.
Compatibility
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
Manufacturer states, “Should not be mixed with other drugs unless specific compatibility data are available,” and lists as incompatible with heparin. Prolonged contact with solutions of an alkaline pH (e.g., sodium lactate, sodium bicarbonate) will result in degradation of idarubicin.
One source suggests the following compatibilities:
Y-site:
Amifostine (Ethyol), amikacin, aztreonam (Azactam), cladribine (Leustatin), cyclophosphamide (Cytoxan), cytarabine (ARA-C), diphenhydramine (Benadryl), droperidol (Inapsine), erythromycin (Erythrocin), etoposide phosphate (Etopophos), filgrastim (Neupogen), gemcitabine (Gemzar), granisetron (Kytril), imipenem-cilastatin (Primaxin), magnesium sulfate, mannitol, melphalan (Alkeran), metoclopramide (Reglan), potassium chloride (KCl), ranitidine (Zantac), sargramostim (Leukine), thiotepa, vinorelbine (Navelbine).
Rate of administration
A single dose of properly diluted medication over 10 to 15 minutes through Y-tube or three-way stopcock of a free-flowing infusion of D5W or NS.
Actions
A highly toxic, synthetic, antibiotic, antineoplastic agent. An analog of daunorubicin. Rapidly distributed; has an increased rate of cellular uptake compared to other anthracyclines. It inhibits synthesis of DNA and interacts with the enzyme topoisomerase II. Results in a greater number of remissions and longer survival than previous protocols (daunorubicin and cytarabine). It is severely immunosuppressive. Extensive extrahepatic metabolism. Half-life averages 20 to 22 hours. Slowly excreted in bile and urine.
Indications and uses
Treatment of acute myeloid leukemia (AML) in adults in combination with other approved antileukemic drugs.
Unlabeled uses:
Treatment of acute lymphoblastic leukemia in pediatric patients; see Maternal/Child.
Contraindications
Not absolute; pre-existing bone marrow suppression, impaired cardiac function, preexisting infection; see Precautions/Monitor. ■ Do not administer if bilirubin above 5 mg/dL.
Precautions
Follow guidelines for handling cytotoxic agents. See Appendix A, p. 1331. ■ Administered by or under the direction of the physician specialist, with facilities for monitoring the patient and responding to any medical emergency. ■ For IV use only. Do not give IM or SC. ■ Use extreme caution in pre-existing drug-induced bone marrow suppression, existing heart disease, previous treatment with other anthracyclines (e.g., daunorubicin), other cardiotoxic agents (e.g., bleomycin), or radiation therapy encompassing the heart. ■ Myocardial toxicity may cause potentially fatal acute congestive heart failure, acute life-threatening arrhythmias, or other cardiomyopathies. Cardiac toxicity is more common in patients who have received prior anthracyclines (e.g., doxorubicin) or who have pre-existing cardiac disease. ■ May cause severe myelosuppression. ■ Use with caution in patients with hepatic or renal dysfunction. Metabolism and excretion of idarubicin may be impaired; see Dose Adjustments.
Monitor:
Determine absolute patency of vein. A stinging or burning sensation indicates extravasation, but extravasation may occur without stinging or burning; severe cellulitis and tissue necrosis can occur with extravasation. Discontinue injection; use another vein. ■ Monitoring of WBCs, RBCs, platelet count, liver function, kidney function, ECG, chest x-ray, echocardiography, and systolic ejection fraction indicated before and during therapy. ■ Severe myelosuppression occurs with effective therapeutic doses. Observe closely for all signs of infection or bleeding. ■ Monitor for thrombocytopenia (platelet count less than 50,000/mm3). Initiate precautions to prevent excessive bleeding (e.g., inspect IV sites, skin, and mucous membranes; use extreme care during invasive procedures; test urine, emesis, stool, and secretions for occult blood). ■ Prophylactic antibiotics may be indicated pending results of C/S in a febrile neutropenic patient. ■ Prophylactic antiemetics may reduce nausea and vomiting and increase patient comfort. ■ Monitor uric acid levels; maintain hydration; allopurinol and urine alkalinization may be indicated. ■ See Precautions.
Patient education:
Nonhormonal birth control recommended. ■ Report IV site burning or stinging promptly. ■ See Appendix D, p. 1333.
Maternal/child:
Category D: avoid pregnancy. May produce teratogenic effects on the fetus. Contraceptive measures indicated during childbearing years. ■ Discontinue breast-feeding before taking idarubicin. ■ Safety and efficacy for use in pediatric patients not established but has been used; consult literature.
Elderly:
Cardiotoxicity or myelotoxicity may be more severe. Patients over 60 years of age who were undergoing induction experienced CHF, serious arrhythmias, chest pain, myocardial infarction, and asymptomatic declines in left ventricular ejection fraction more frequently than younger patients. ■ Monitor renal, hepatic, and hematologic functions closely.
Drug/lab interactions
Bone marrow toxicity is additive with other chemotherapeutic agents. ■ Risk of cardiotoxicity increased in patients previously treated with maximum cumulative doses of other anthracyclines (e.g., doxorubicin [Adriamycin], mitoxantrone [Novantrone]) and/or radiation encompassing the heart. ■ Leukopenic and/or thrombocytopenic effects may be increased with drugs that cause blood dyscrasias (e.g., anticonvulsants [e.g., carbamazepine (Tegretol), phenytoin (Dilantin)], NSAIDs [e.g., ibuprofen (Advil, Motrin), naproxen (Aleve, Naprosyn)]). Adjust dose based on differential and platelet count. ■ Do not administer live virus vaccines to patients receiving antineoplastic drugs. ■ See Precautions/Monitor.
Side effects
Acute congestive heart failure, alopecia (reversible), arrhythmias, bone marrow suppression (marked with average doses), cramping, decrease in systolic ejection fraction, depressed QRS voltage, diarrhea, erythema and tissue necrosis (if extravasation occurs), fever, headache, hemorrhage (severe), hepatic function changes, infection, mucositis, myocarditis, nausea, pericarditis, renal function changes, seizures, skin rash, urticaria (local), vomiting.
Antidote
Most side effects will be tolerated or treated symptomatically. Keep physician informed. Close monitoring of bone marrow, ECG, chest x-ray, echocardiography, and systolic ejection fraction may prevent most serious and potentially fatal side effects. There is no specific antidote, but adequate supportive care including platelet transfusions, antibiotics, and symptomatic treatment of mucositis is required. For extravasation, elevate the extremity and apply intermittent ice packs over the area immediately and 4 times a day for 1/2 hour. Continue for 3 days. Consider aspiration of as much infiltrated drug as possible, flooding of the site with NS, and injection of hydrocortisone sodium succinate (Solu-Cortef) throughout extravasated tissue. Use a 27- or 25-gauge needle. Site should be observed promptly by a reconstructive surgeon. If ulceration begins or there is severe persistent pain at the site, early wide excision of the involved area will be considered. Hemodialysis or peritoneal dialysis probably not effective in overdose.
Idarucizumab
(EYE-da-roo-KIZ-ue-mab)
Praxbind
Antidote
Monoclonal antibody
pH 5.3 to 5.7
Usual dose
5 Gm provided as two separate vials, each containing 2.5 Gm/50 mL. There are limited data to support administration of an additional 5-Gm dose of idarucizumab; see Precautions.
Dose adjustments
No dose adjustment is required in renally impaired patients. ■ Formal studies in patients with hepatic impairment have not been conducted.
Dilution
A colorless to slightly yellow, clear to slightly opalescent solution available as a 2.5 Gm/50 mL injection in a single-use vial. Administer undiluted by withdrawing and injecting the contents of 2 vials with a syringe or by hanging and infusing the contents of 2 vials.
Filter:
Specific information not available.
Storage:
Store unopened vials in the refrigerator at 2° to 8° C (36° to 46° F). Do not freeze. Do not shake. Before use, the unopened vial may be kept at RT 25° C (77° F) for up to 48 hours if stored in the original package to protect from light, or up to 6 hours when exposed to light. Once solution has been removed from the vial, administration should begin promptly or within 1 hour.
Compatibility
Manufacturer states, “Do not mix with other medicinal products. A pre-existing IV line may be used for administration. The line must be flushed with NS prior to infusion. No other infusion should be administered in parallel via the same IV access.”
Rate of administration
Administer by bolus injection by injecting both vials consecutively one after another via syringe. Alternatively, may administer as two rapid consecutive infusions by hanging and infusing each vial one after another. Flush IV line with NS before administration of idarucizumab.
Actions
A recombinant humanized monoclonal antibody fragment (Fab); it is derived from an IgG1 isotype molecule whose target is the direct thrombin inhibitor dabigatran. Idarucizumab is a specific reversal agent for dabigatran. It binds to dabigatran and its acyl glucuronide metabolites with a higher affinity than the binding affinity of dabigatran to thrombin, thereby neutralizing their anticoagulant effect. In studies, the plasma concentrations of unbound dabigatran were reduced to below the lower limit of quantification immediately after the administration of 5 Gm idarucizumab. Subjects’ diluted thrombin time (dTT), ecarin clotting time (ECT), activated partial thromboplastin time (aPTT), thrombin time (TT), and activated clotting time (ACT) parameters returned to baseline levels. Idarucizumab is rapidly eliminated with an initial half-life of 47 minutes and a terminal half-life of 10.3 hours. Some drug is recovered in the urine. The remaining part of the dose is assumed to be eliminated via protein catabolism, mainly in the kidney. Idarucizumab is thought to be metabolized via pathways that involve the biodegradation of the antibody to smaller peptides or amino acids, which are then reabsorbed and incorporated in the general protein synthesis.
Indications and uses
Reversal of the anticoagulant effects of dabigatran (Pradaxa) when reversal is needed for emergency surgery/urgent procedures or in life-threatening or uncontrolled bleeding.
Limitations of use:
This indication is approved under accelerated approval based on a reduction in unbound dabigatran and normalization of coagulation parameters in healthy volunteers. Continued approval for this indication may be contingent on the results of an ongoing cohort case series study.
Contraindications
Manufacturer states, “None.”
Precautions
For IV use only. ■ Idarucizumab treatment should be used in conjunction with standard supportive measures. ■ Reversing dabigatran therapy exposes patients to the thrombotic risk of their underlying disease. To reduce this risk, resumption of anticoagulant therapy should be considered as soon as medically appropriate. Dabigatran treatment can be initiated 24 hours after administration of idarucizumab. No changes in the pharmacokinetics or pharmacodynamics of dabigatran were noted upon re-initiation 24 hours after administration of idarucizumab. ■ Elevated coagulation parameters (e.g., activated partial thromboplastin time [aPPT], ecarin clotting time [ECT]) have been observed 12 and 24 hours after administration of a 5-Gm dose of idarucizumab in a limited number of patients. Administration of an additional 5-Gm dose of idarucizumab may be considered if the reappearance of clinically relevant bleeding together with elevated coagulation parameters is observed after administration of the initial dose. Similarly, patients who require a second emergency surgery/urgent procedure and have elevated coagulation parameters may receive an additional 5-Gm dose of idarucizumab. The safety and effectiveness of repeat treatment with idarucizumab have not been established. ■ Adverse events possibly indicative of hypersensitivity reactions have been reported. The risk of using idarucizumab in a patient with known hypersensitivity to idarucizumab (e.g., anaphylactic reaction) or to any of its excipients needs to be weighed cautiously against the potential benefit of such an emergency treatment. If an anaphylactic reaction or other serious reaction occurs, immediately discontinue administration of idarucizumab. ■ The recommended dose of idarucizumab contains 4 Gm of sorbitol as an excipient. Patients with the condition of hereditary fructose intolerance (HFI) have experienced serious adverse reactions (e.g., acute liver failure, hypoglycemia, hypophosphatemia, increase in uric acid, metabolic acidosis) after receiving parenteral administration of sorbitol. When prescribing idarucizumab to patients with HFI, consider the combined daily metabolic load of sorbitol/fructose from all sources, including idarucizumab and other drugs containing sorbitol. The minimum amount of sorbitol at which serious adverse reactions may occur in these patients is not known. ■ Idarucizumab is a specific reversal agent for dabigatran, with no impact on the effect of other anticoagulant or antithrombotic therapies. ■ As with all proteins, there is a potential for immunogenicity with idarucizumab. Pre-existing antibodies with cross-reactivity to idarucizumab were detected in some subjects. No impact on the pharmacokinetics or the reversal effect of idarucizumab or on hypersensitivity reactions was observed in these subjects.
Monitor:
Monitor coagulation parameters (e.g., aPPT and ECT [if available]). ■ Monitor for S/S of clinically relevant bleeding and thromboembolic events. ■ Monitor for S/S of hypersensitivity reactions (e.g., bronchospasm, fever, pruritus, rash).
Patient education:
Increased thromboembolic risk with reversal of dabigatran. Resume anticoagulant therapy as directed when deemed stable. ■ Seek immediate medical attention for any S/S of bleeding. ■ Seek immediate medical attention for S/S of a hypersensitivity reaction (e.g., rash, wheezing). Reactions may occur during or after administration of idarucizumab. ■ Inform patients with HFI that idarucizumab contains sorbitol. Adverse reactions related to the parenteral administration of sorbitol may occur during or after administration and should be promptly reported.
Maternal/child:
Risk to fetus unknown. Administer during pregnancy only if clearly needed. ■ Safety and effectiveness during labor and delivery have not been studied. ■ Use caution during breast-feeding; risk to infant unknown. ■ Safety and effectiveness have not been established in pediatric patients.
Elderly:
No overall differences in safety or effectiveness have been observed between elderly patients and younger patients.
Drug/lab interactions
Formal drug interaction studies have not been conducted. ■ In vitro data suggest that the inhibition of dabigatran by idarucizumab is not affected by coagulation factor concentrates (3-factor or 4-factor prothrombin complex concentrates [PCCs], activated PCC, or recombinant factor VIIa).
Side effects
The most frequently reported adverse reactions were constipation, delirium, fever, headache, hypokalemia, and pneumonia. Hypersensitivity reactions (e.g., bronchospasm, fever, hyperventilation, pruritus, and rash) and thromboembolic events have been reported.
Antidote
Notify physician of any side effects; most will be treated symptomatically. Discontinue administration at the first sign of a serious hypersensitivity reaction and treat as indicated (e.g., oxygen, diphenhydramine, epinephrine, corticosteroids, vasopressors, and/or fluids). Resuscitate as necessary.
Ifosfamide 
(eye-FOS-fah-myd)
Ifex, Ifosfamide/Mesna Kit
Antineoplastic (alkylating agent/nitrogen mustard)
pH 6
Usual dose
Specific testing recommended before each dose; see Monitor.
1.2 Gm/M2/day for 5 consecutive days. Repeat every 3 weeks as hematologic recovery permits. To prevent hemorrhagic cystitis, ifosfamide should be given with extensive hydration (e.g., at least 2 liters of oral or IV fluid/day), and mesna should be administered with every dose. Ifosfamide dose has been mixed with the initial mesna dose each day in the same solution. Appears to be compatible.
Dose adjustments
Reduced dose may be required in renal or hepatic impairment. Adequate data not available. ■ Dosing should be cautious in the elderly. Consider decrease in cardiac, hepatic, and renal function; concomitant disease; or other drug therapy. See Elderly. ■ Severe myelosuppression is frequent, especially when ifosfamide is given with other chemotherapeutic agents. Dose adjustments of all agents may be required. Adjustment of dose interval may be required. Unless clinically essential, the dose should be held for WBCs less than 2,000/mm3 and/or platelets less than 50,000/mm3. See Monitor and Drug/Lab Interactions.
Dilution
Specific techniques required; see precautions.
Each 1 Gm must be diluted with 20 mL SWFI or bacteriostatic water for injection (parabens or benzyl alcohol preserved only). Shake solution to dissolve. May be further diluted with D5W, NS, LR, or SWFI to a final concentration of 0.6 to 20 mg/mL. Further dilution with D21/2W, 1/2NS, and D5NS is also acceptable for larger volumes. 1 Gm in 20 mL or 3 Gm in 60 mL equals 50 mg/mL; 1 Gm in 50 mL equals 20 mg/mL; 1 Gm in 200 mL equals 5 mg/mL. Generic product available in a kit containing ifosfamide and mesna.
Filters:
Studies measured potency of ifosfamide in combination with mesna through a 5-micron filter. No significant drug loss for ifosfamide; mesna was not measured.
Storage:
Store at CRT 20° to 25° C (68° to 77° F). Protect from temperatures above 30° C (86° F). Reconstituted and further diluted solution should be refrigerated and used within 24 hours. Solutions containing benzyl alcohol can reduce stability.
Compatibility (underline indicates conflicting compatibility information)
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
One source suggests the following compatibilities:
Additive:
Carboplatin (Paraplatin), cisplatin, epirubicin (Ellence), etoposide (VePesid), fluorouracil (5-FU), mesna (Mesnex).
Y-site:
Allopurinol (Aloprim), amifostine (Ethyol), amphotericin B cholesteryl (Amphotec), anidulafungin (Eraxis), aztreonam (Azactam), caspofungin (Cancidas), doripenem (Doribax), doxorubicin liposomal (Doxil), etoposide phosphate (Etopophos), filgrastim (Neupogen), fludarabine (Fludara), gallium nitrate (Ganite), gemcitabine (Gemzar), granisetron (Kytril), linezolid (Zyvox), melphalan (Alkeran), ondansetron (Zofran), oxaliplatin (Eloxatin), paclitaxel (Taxol), palonosetron (Aloxi), pemetrexed (Alimta), piperacillin/tazobactam (Zosyn), propofol (Diprivan), sargramostim (Leukine), sodium bicarbonate, teniposide (Vumon), thiotepa, topotecan (Hycamtin), vinorelbine (Navelbine).
Rate of administration
A single dose over a minimum of 30 minutes as an infusion. Extend administration time based on amount of diluent and patient condition.
Actions
An alkylating agent. Chemically related to the nitrogen mustards and a synthetic analog of cyclophosphamide. A prodrug that requires activation by hepatic cytochrome P450 isoenzymes to exert its cytotoxic activity. Exact mechanism of action not determined, but its cytotoxic action is primarily through DNA cross-links caused by alkylation by isophosphoramide mustard at guanine N7 positions. The formation of inter- and intra-strand cross-links in the DNA results in cell death. Distribution takes place with minimal tissue binding and little plasma protein binding. Extensively bound by red blood cells. Extensively metabolized in the liver by cytochrome P450 isoenzymes (considerable individual variation). Elimination half-life for a usual dose is about 7 hours. Half-life is extended with larger doses. Ifosfamide and its metabolites are excreted in urine. Secreted in breast milk.
Indications and uses
Used in combination with other specific antineoplastic agents for third-line chemotherapy of germ cell testicular cancer. Should be used in combination with mesna for prophylaxis of hemorrhagic cystitis.
Unlabeled uses:
Bladder, cervical, and ovarian cancers; Ewing’s sarcoma; Hodgkin’s and non-Hodgkin’s lymphoma; small-cell lung cancer; osteosarcoma; soft tissue sarcomas.
Contraindications
Hypersensitivity to ifosfamide; urinary outflow obstruction.
Precautions
Follow guidelines for handling cytotoxic agents. See Appendix A, p. 1331. ■ Usually administered by or under the direction of the physician specialist. ■ Exclude or correct any urinary tract obstructions; see Contraindications. ■ Administer cautiously, if at all, to patients with an infection (including an active urinary tract infection), severe immunosuppression, or compromised bone marrow reserve as indicated by leukopenia, granulocytopenia, extensive bone marrow metastases, prior radiation therapy, or prior treatment with other cytotoxic agents. ■ Myelosuppression can be severe and lead to fatal infections. Deaths have been reported. Risk of myelosuppression is dose-dependent and is increased in patients with reduced renal function and when ifosfamide is administered as a single high dose compared to fractionated doses. Severe myelosuppression is often observed when ifosfamide is used in combination with other chemotherapeutic/hematotoxic agents and/or radiation therapy. ■ CNS toxicities can be severe and result in encephalopathy and death. ■ Nephrotoxic and urotoxic. Acute tubular necrosis, acute renal failure, and chronic renal failure have been reported, and deaths have occurred. Benefits must outweigh risks if ifosfamide is used in patients with pre-existing renal impairment or reduced nephron reserve. Glomerular and tubular disorders of renal function are common and may become apparent during therapy or months and years after stopping treatment. They may resolve, remain stable, or progress even after completion of treatment. Hemorrhagic cystitis can be severe and may require blood transfusions. Urotoxic effects can be reduced by the prophylactic use of mesna. Risk of hemorrhagic cystitis is dose-dependent and increases with the administration of single high doses compared to fractionated administration. Past or concomitant radiation of the bladder or busulfan therapy may also increase risk. ■ Use caution in patients with risk factors for cardiotoxicity or pre-existing cardiac disease. Cardiotoxicity (e.g., supraventricular or ventricular arrhythmias, decreased QRS voltage and ST-segment or T-wave changes, toxic cardiomyopathy leading to heart failure with congestion and hypotension, pericardial effusion, fibrinous pericarditis, epicardial fibrosis), sometimes with fatal outcomes, has been reported. Risk is dose dependent and is increased with prior or concomitant treatment with other cardiotoxic agents or radiation of the cardiac region and, possibly, renal impairment. ■ Pulmonary toxicity (e.g., interstitial pneumonitis, pulmonary fibrosis, and other forms of pulmonary toxicity) has been reported, sometimes with fatal outcomes. ■ Hypersensitivity reactions, including anaphylaxis, have been reported. Cross-sensitivity between oxazaphosphorine cytotoxic agents (e.g., cyclophosphamide [Cytoxan]) has been reported. ■ May interfere with normal wound healing. ■ Risk of secondary malignancies has been reported. ■ Veno-occlusive liver disease has been reported. ■ Studies in patients with hepatic or renal impairment have not been conducted. Use with caution.
Monitor:
Monitor blood counts prior to and at intervals after each treatment cycle. CBC with differential and platelet count and hemoglobin are recommended before each daily dose and as clinically indicated. Unless clinically essential, WBC count should be above 2,000/mm3 and platelet count above 50,000/mm3. ■ Evaluate glomerular and tubular kidney function before, during, and after treatment. ■ Monitor urinary sediment regularly for erythrocytes and other signs of urotoxicity/nephrotoxicity. ■ Urinalysis before each dose recommended. Withhold drug if RBCs in urine exceed 10 per high-powered field. Reinstitute after complete resolution with vigorous oral or parenteral hydration. Mesna given concurrently should prevent hemorrhagic cystitis. ■ Monitor serum and urine chemistries, including phosphorus and potassium, regularly; administer replacement as indicated. ■ Monitor for S/S of renal dysfunction (e.g., decreased glomerular filtration rate, increased SCr, aminoaciduria, cylindruria, enzymuria, glycosuria, phosphaturia, proteinuria, and tubular acidosis). ■ Adequate hydration required; encourage fluid intake (minimum of 2 L/day) and frequent voiding to prevent cystitis. ■ Nadir of the leukocyte count tends to be reached during the second week after administration. ■ Monitor for S/S of hypersensitivity (e.g., chest pain, dizziness, dyspnea, fever, flushing, hypotension, nausea, pruritus, rash, rigors, urticaria). ■ Monitor for S/S of pulmonary toxicity and treat as indicated. ■ Pneumonias, as well as other bacterial, fungal, viral, and parasitic infections, have been reported, and latent infections can be reactivated. Observe constantly for signs of infection (e.g., fever, sore throat, tiredness) or unusual bleeding or bruising. Antimicrobial prophylaxis may be indicated in certain cases of neutropenia. Antibiotics and/or antimycotics must be given if neutropenic fever occurs. ■ Monitor for thrombocytopenia (platelet count less than 50,000/mm3). Initiate precautions to prevent excessive bleeding (e.g., inspect IV sites, skin, and mucous membranes; use extreme care during invasive procedures; test urine, emesis, stool, and secretions for occult blood). ■ Monitor for CNS toxicity. Blurred vision, coma, confusion, extrapyramidal symptoms, hallucinations, peripheral neuropathy, psychotic behavior, seizures, somnolence, and urinary incontinence have been reported. Neurotoxicity may appear within a few hours to a few days after the first administration and usually resolves within 48 to 72 hours of discontinuing ifosfamide, although symptoms may persist. Recurrence after several uneventful treatment courses has occurred. Discontinue treatment if encephalopathy occurs. ■ Prophylactic administration of antiemetics recommended. ■ See Drug/Lab Interactions.
Patient education:
Nonhormonal birth control recommended. Females should avoid pregnancy and males should not father a child during therapy and for up to 6 months after the end of therapy; see Maternal/Child. ■ CNS toxicity may impair the ability to operate an automobile or other heavy machinery. ■ Side effects are numerous and may be fatal; review all potential side effects with your health care professional before treatment. ■ See Appendix D, p. 1333.
Maternal/child:
Category D: can cause fetal harm. Females should avoid pregnancy and males should not father a child during therapy and for up to 6 months after the end of therapy. May cause amenorrhea in females and oligospermia or azoospermia in males. Sterility has been reported. ■ Discontinue breast-feeding. ■ Safety and effectiveness for use in pediatric patients not established. Fanconi syndrome, renal rickets, and growth retardation have been reported in pediatric patients. Prepubescent females may become sterile or are at risk for developing premature menopause. Prepubescent males may not develop secondary sexual characteristics normally and may have oligospermia or azoospermia.
Elderly:
Consider age-related organ impairment. Dose selection should be cautious; see Dose Adjustments. Monitor renal, hepatic, and hematologic functions closely.
Drug/lab interactions
See Dose Adjustments. ■ A substrate for both CYP3A4 and CYP2B6. CYP3A4 inducers (e.g., carbamazepine [Tegretol], fosphenytoin [Cerebyx], phenytoin [Dilantin], phenobarbital [Luminal], rifampin [Rifadin], and St. John’s wort) may increase the metabolism of ifosfamide to its active alkylating metabolites and increase the formation of the neurotoxic/nephrotoxic metabolite chloroacetaldehyde. Monitor closely for signs of toxicity; consider dose adjustment. CYP3A4 inhibitors (e.g., aprepitant [Emend], fosaprepitant [Emend for injection], fluconazole [Diflucan], grapefruit and grapefruit juice, itraconazole [Sporanox], ketoconazole [Nizoral], sorafenib [Nexavar]) may decrease the metabolism of ifosfamide to its active alkylating metabolites, perhaps decreasing the effectiveness of treatment. ■ May have additive effects with other drugs that act on the CNS (e.g., antihistamines, antiemetics, narcotics, or sedatives). Use with caution or discontinue if encephalopathy occurs. ■ Do not administer live virus vaccines to patients receiving antineoplastic drugs.
Side effects
Hematuria, hemorrhagic cystitis, and bone marrow suppression are dose-limiting side effects. The most common side effects included alopecia, anemia, CNS toxicity, hematuria, infection, leukopenia, nausea, and vomiting. Other commonly reported side effects were anorexia, confusion, constipation, depressive psychosis with hallucinations, diarrhea, and somnolence. Cardiotoxicity, coagulopathy, coma, cranial nerve dysfunction, dermatitis, disorientation, dizziness, fatigue, fever of unknown origin, hypersensitivity reactions, hypertension, hypotension, liver dysfunction, malaise, neutropenia, osteomalacia, phlebitis, polyneuropathy, pulmonary symptoms, thrombocytopenia, or seizures may occur.
Overdose:
Manifestations of dose-dependent toxicities such as CNS toxicity, mucositis, myelosuppression, nephrotoxicity.
Post-marketing:
Anaphylaxis; angioedema; benign and malignant neoplasms; hematotoxicity (e.g., agranulocytosis, bone marrow failure, DIC, febrile bone marrow aplasia, hemolytic anemia, hemolytic uremic syndrome, methemoglobinemia, neonatal anemia); infections, including reactivation of latent infections (e.g., herpes zoster, Pneumocystis jiroveci, progressive multifocal leukoencephalopathy [PML], Strongyloides, viral hepatitis, and other viral and fungal infections); pneumonia; sepsis and septic shock (including fatal outcomes); psychiatric disorders; syndrome of inappropriate antidiuretic hormone secretion (SIADH); tumor lysis syndrome; and numerous other serious side effects have been reported.
Antidote
Minor side effects will be treated symptomatically if necessary. Discontinue ifosfamide and notify physician immediately if hematuria, hemorrhagic cystitis, confusion, coma, WBC below 2,000 mm3, or platelets below 50,000/mm3 occur. Administration of whole blood products (e.g., packed RBCs, platelets, leukocytes) and/or blood modifiers (e.g., darbepoetin alfa [Aranesp], epoetin alfa [Epogen], filgrastim [Neupogen, Zarxio], pegfilgrastim [Neulasta], sargramostim [Leukine]) may be indicated to treat bone marrow suppression. Maintain supportive therapy for CNS symptoms until complete resolution; occasionally, recovery has been incomplete. Discontinue treatment if encephalopathy occurs. There is no specific antidote. Supportive therapy as indicated will help sustain the patient in toxicity. Cystitis prophylaxis with mesna may prevent or limit urotoxic effects. Ifosfamide and its metabolites are dialyzable.
Imipenem-cilastatin
(em-ee-PEN-em sigh-lah-STAT-in)
Primaxin
Antibacterial (carbapenem)
pH 6.5 to 8.5
Usual dose
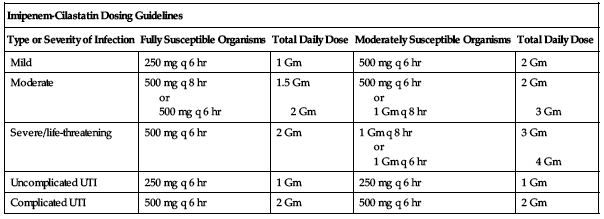
Range is from 250 mg to 1 Gm every 6 to 8 hours. Dose based on severity of disease, susceptibility of pathogens, condition of the patient, age, weight, and CrCl. Adult doses in the following chart are limited to patients with a CrCl equal to or greater than 71 mL/min/1.73 M2 and a body weight equal to or greater than 70 kg; see Dose Adjustments. Because of high antimicrobial activity, do not exceed the lower of 50 mg/kg/24 hr or 4 Gm/24 hr in adult or pediatric patients. Continue for at least 2 days after all symptoms of infection subside.
| Imipenem-Cilastatin Dosing Guidelines | ||||
| Type or Severity of Infection | Fully Susceptible Organisms | Total Daily Dose | Moderately Susceptible Organisms | Total Daily Dose |
| Mild | 250 mg q 6 hr | 1 Gm | 500 mg q 6 hr | 2 Gm |
| Moderate | 500 mg q 8 hr or 500 mg q 6 hr | 1.5 Gm 2 Gm | 500 mg q 6 hr or 1 Gm q 8 hr | 2 Gm 3 Gm |
| Severe/life-threatening | 500 mg q 6 hr | 2 Gm | 1 Gm q 8 hr or 1 Gm q 6 hr | 3 Gm 4 Gm |
| Uncomplicated UTI | 250 mg q 6 hr | 1 Gm | 250 mg q 6 hr | 1 Gm |
| Complicated UTI | 500 mg q 6 hr | 2 Gm | 500 mg q 6 hr | 2 Gm |
Adult and pediatric patients over 12 years of age with cystic fibrosis and normal renal function:
Higher doses up to 90 mg/kg/day have been used. Do not exceed 4 Gm/24 hr.
Pediatric dose
Not recommended for use in pediatric patients with CNS infections because of the risk of seizures, or in pediatric patients weighing less than 30 kg with impaired renal function (no data available). Because of high antimicrobial activity, do not exceed the lower of 50 mg/kg/24 hr or 4 Gm/24 hr in adult or pediatric patients.
Infants 3 months of age or less, weighing 1,500 gm or more: For non-CNS infections:
Less than 1 week of age: 25 mg/kg every 12 hours.
1 to 4 weeks of age: 25 mg/kg every 8 hours.
4 weeks to 3 months of age: 25 mg/kg every 6 hours.
Premature infants weighing from 670 to 1,890 Gm in the first week of life were given 20 mg/kg every 12 hours in one study (see package insert).
Infants and other pediatric patients over 3 months of age: For non-CNS infections: 15 to 25 mg/kg/dose every 6 hours. See statements in Usual Dose. Maximum dose is 2 Gm/day for treatment of infections caused by fully susceptible organisms and 4 Gm/day for moderately susceptible organisms.
Dose adjustments
Dose adjustments are extensive and required for all patients weighing less than 70 kg, as well as all patients with a CrCl less than 71 mL/min/1.73 M2. Dose adjustments also vary based on total daily dose required (e.g., 1, 1.5, 2, 3, or 4 Gm). See charts in package insert. ■ Reduced doses may be required in the elderly based on weight and/or decreased renal function. ■ Cleared by hemodialysis; administer after hemodialysis and at 12-hour intervals thereafter. ■ See Precautions/Monitor and Drug/Lab Interactions.
Dilution
Reconstitute each single dose with 10 mL of compatible infusion solutions (e.g., D5W, D10W, NS [see chart on inside back cover or literature]); also compatible in D5W with 0.15% KCl and mannitol 5% and 10%. Shake well and transfer the resulting suspension to 100 mL of the same infusion solution. Rinse vial with an additional 10 mL of infusion solution to ensure complete transfer of vial contents to the infusion solution. Agitate until clear. Also available in ADD-Vantage vials for use with ADD-Vantage infusion containers.
Neonatal dilution:
Use preservative-free solutions for reconstitution of neonatal doses.
Filters:
Manufacturer’s data limited; indicates that use of a filter system when withdrawing the suspension from the vial would probably result in filter clogging and decrease the available antibiotic. An in-house study documented compatibility of the final diluted solution with a 0.22-micron in-line filter.
Storage:
Store dry powder below 25° C (77° F). Diluted solutions are stable at room temperature for 4 hours after preparation or 24 hours if refrigerated. Do not freeze.
Compatibility (underline indicates conflicting compatibility information)
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
Manufacturer states, “Should not be mixed with or physically added to other antibiotics.” See Drug/Lab Interactions.
One source suggests the following compatibilities:
Y-site:
Acyclovir (Zovirax), amifostine (Ethyol), anidulafungin (Eraxis), aztreonam (Azactam), caspofungin (Cancidas), cisatracurium (Nimbex), diltiazem (Cardizem), docetaxel (Taxotere), famotidine (Pepcid IV), filgrastim (Neupogen), fludarabine (Fludara), foscarnet (Foscavir), granisetron (Kytril), idarubicin (Idamycin), insulin (regular), linezolid (Zyvox), melphalan (Alkeran), methotrexate, ondansetron (Zofran), propofol (Diprivan), remifentanil (Ultiva), tacrolimus (Prograf), telavancin (Vibativ), teniposide (Vumon), thiotepa, tigecycline (Tygacil), vasopressin, vinorelbine (Navelbine), zidovudine (AZT, Retrovir).
Rate of administration
Intermittent IV:
Each 500 mg or fraction thereof over 20 to 30 minutes. Doses greater than 500 mg should be infused over 40 to 60 minutes. Slow infusion rate if patient develops nausea and vomiting, dizziness, hypotension, or sweating. May be given through Y-tube or three-way stopcock of infusion set.
Actions
A potent broad-spectrum antibacterial agent. Imipenem is a carbapenem antibiotic; cilastatin inhibits the kidney enzyme responsible for the metabolism of imipenem. Both components are present in equal amounts. Bactericidal to many gram-negative, gram-positive, and anaerobic organisms. Bactericidal activity results from the inhibition of bacterial wall synthesis. Effective against many otherwise resistant organisms. Has a high degree of stability in the presence of beta-lactamases produced by gram-negative and gram-positive bacteria. Rapidly and widely distributed into many body fluids and tissues. Metabolized in the kidneys. Half-life is approximately 1 hour. Excreted in the urine. May cross the placental barrier.
Indications and uses
Treatment of serious lower respiratory tract, urinary tract, skin and skin structure, bone and joint, gynecologic, intra-abdominal, and polymicrobic infections; bacterial septicemia; and endocarditis. Most effective against specific organisms (see literature).
Unlabeled uses:
Treatment of febrile neutropenia and melioidosis (a rare infection in humans and animals).
Contraindications
Known sensitivity to any component of this product. ■ See Maternal/Child and Precautions.
Precautions
Specific sensitivity studies are indicated to determine susceptibility of the causative organism to imipenem-cilastatin. ■ To reduce the development of drug-resistant bacteria and maintain its effectiveness, imipenem-cilastatin should be used to treat or prevent only those infections proven or strongly suspected to be caused by bacteria. ■ Serious and occasionally fatal hypersensitivity reactions have been reported in patients receiving therapy with beta-lactams (e.g., carbapenems, cephalosporins, penicillins). More likely in patients with a history of sensitivity to multiple allergens; obtain a careful history. Cross-sensitivity is possible. ■ Avoid prolonged use of drug; superinfection caused by overgrowth of nonsusceptible organisms may result. ■ CNS adverse effects, including confusional states, myoclonic activity, and seizures, have been reported. Incidence increases with higher doses, compromised renal function, or pre-existing CNS disorders. ■ Not recommended in patients with a CrCl less than 5 mL/min/1.73 M2 unless hemodialysis is instituted within 48 hours. ■ For patients on hemodialysis, benefits of use must outweigh risk of seizures. ■ Clostridium difficile–associated diarrhea (CDAD) has been reported. May range from mild diarrhea to fatal colitis. Consider in patients who present with diarrhea during or after treatment with imipenem-cilastatin. ■ Safety and effectiveness for use in meningitis not established. ■ Clinical improvement has been observed in patients with cystic fibrosis, COPD, and lower respiratory tract infections caused by Pseudomonas aeruginosa; however, effective bacterial eradication may not occur. ■ See Maternal/Child and Drug/Lab Interactions.
Monitor:
May cause thrombophlebitis. Use small needles and large veins, and rotate infusion sites. ■ Electrolyte imbalance and cardiac irregularities resulting from sodium content are possible. Contains 3.2 mEq of sodium/Gm. ■ Monitor renal, hepatic, and hemopoietic systems in prolonged therapy. ■ Some strains of Pseudomonas aeruginosa may develop resistance fairly rapidly during treatment. Periodic susceptibility testing should be done when clinically indicated. ■ See Drug/Lab Interactions.
Patient education:
Promptly report diarrhea or bloody stools that occur during treatment or up to several months after an antibiotic has been discontinued; may indicate CDAD and require treatment. ■ Patients with a history of seizures should review medication profile with physician before taking imipenem; see Drug/Lab Interactions.
Maternal/child:
Category C: use only if potential benefit outweighs potential risk during pregnancy and breast-feeding.
Elderly:
Consider weight and age-related renal impairment. See Dose Adjustments. ■ Response similar to that seen in younger patients; however, greater sensitivity in the elderly cannot be ruled out.
Drug/lab interactions
Carbapenems may reduce serum valproic acid concentrations to subtherapeutic levels, resulting in loss of seizure control. Monitor valproic acid levels. Consider alternative antibacterial therapy. If administration of imipenem is necessary, supplemental anticonvulsant therapy should be considered. ■ May be used concomitantly with aminoglycosides and other antibiotics, but these drugs must never be mixed in the same infusion or given concurrently. ■ Use with ganciclovir (Cytovene) may cause generalized seizures. Use only if benefit outweighs risk. ■ Half-life and plasma levels slightly increased by probenecid. Avoid concurrent use. ■ Concurrent use with cyclosporine (Sandimmune) may decrease cyclosporine metabolism. Elevated cyclosporine levels and neurotoxicity (e.g., agitation, confusion, tremor) have been reported.
Side effects
Full scope of hypersensitivity reactions including anaphylaxis, pruritus, rash, and urticaria. Abdominal pain; abnormal clotting time; agitation; altered CBC and electrolytes; anuria; burning, discomfort, and pain at injection site; CDAD; confusion; diarrhea; dizziness; dyskinesia; dyspnea; elevated alkaline phosphatase, AST, ALT, bilirubin, creatinine, BUN, LDH; fever; gastroenteritis; glossitis; headache; heartburn; hemorrhagic colitis; hepatic failure; hepatitis (including fulminant hepatitis); hyperventilation; hypotension; increased salivation; myoclonus; nausea and vomiting; paresthesia; pharyngeal pain; polyarthralgia; polyuria; positive direct Coombs’ test; presence of WBCs or RBCs, protein casts, bilirubin, or urobilinogen in urine; seizures; somnolence; thrombophlebitis; tinnitus; tongue papillar hypertrophy; transient hearing loss in the hearing impaired; vertigo; and many others.
Antidote
Notify physician of any side effects. Discontinue the drug if indicated. Treat hypersensitivity reactions as indicated; may require epinephrine, airway management, oxygen, IV fluids, antihistamines (e.g., diphenhydramine [Benadryl]), corticosteroids (e.g., hydrocortisone sodium succinate [Solu-Cortef]), and pressor amines (e.g., dopamine). Resuscitate as necessary. Begin anticonvulsants if focal tremors, myoclonus, or seizures occur. If symptoms continue, decrease dose or discontinue the drug. Infusion rate reactions (e.g., dizziness, hypotension, N/V, and sweating) may respond to a decrease in rate of infusion. Mild cases of CDAD may respond to discontinuation of the drug. Treat CDAD with fluids, electrolytes, protein supplements, and oral vancomycin (Vancocin) or metronidazole (Flagyl) as indicated. In severe cases, surgical evaluation may be indicated. Hemodialysis may be useful in overdose.
Immune globulin intravenous 
(im-MUNE GLAW-byoo-lin IV)
Bivigam, Carimune NF, Flebogamma, Gammagard Liquid, Gammagard S/D, Gammaked, Gammaplex, Gamunex-C, IGIV, Octagam, Privigen
Immunizing agent (passive)
pH 4 to 7.2
Usual dose
For all indications, ensure adequate hydration before administration.
Primary immunodeficiency (PID) diseases
Bivigam and gammuplex:
300 to 800 mg/kg as a single-dose IV infusion every 3 to 4 weeks. Monitor clinical response and trough levels to adjust dose as appropriate. Desired trough level of total IgG concentrations is a minimum of 500 mg/dL (target is 600 mg/dL).
Carimune NF:
400 to 800 mg/kg as a single-dose IV infusion. Administer once every 3 to 4 weeks. Use of a 3% solution is recommended for the first infusion in previously untreated agammaglobulinemic or hypogammaglobulinemic patients. Subsequent infusions may be administered at a higher concentration based on patient tolerance.
Flebogamma, gammagard liquid, gammagard S/D, gammaked, gamunex-C, and octagam 5%:
300 to 600 mg/kg as a single-dose IV infusion every 3 to 4 weeks. Individualize dose and interval based on clinical response. If a patient receiving Gammaked, Gamunex-C, or Octagam is routinely receiving a dose of less than 400 mg/kg and is at risk for measles exposure (i.e., traveling to a measles endemic area), administer a dose of at least 400 mg/kg just before the expected measles exposure. If a patient is exposed to measles, administer a dose of 400 mg/kg as soon as possible after exposure.
Privigen:
200 to 800 mg/kg as a single-dose IV infusion every 3 to 4 weeks. Adjust dose to achieve desired serum trough levels and clinical response.
Idiopathic thrombocytopenic purpura (ITP)
All products must be given by IV infusion for ITP patients. Do not administer SC due to a potential risk of hematoma.
Carimune NF:
400 mg/kg/day for 2 to 5 consecutive days based on platelet count and clinical response. A 6% solution is recommended for use in ITP; see chart in Rate of Administration. May be discontinued in acute ITP of childhood if an initial platelet count response to the first 2 doses is adequate (30,000 to 50,000/mm3). After induction, if clinically significant bleeding occurs and/or the platelet count falls below 30,000/mm3, a maintenance dose of 400 mg/kg may be given as a single infusion. If response inadequate, increase to 800 to 1,000 mg/kg. May be given intermittently to maintain platelet count.
Flebogamma 10%:
1 Gm/kg/day for 2 consecutive days.
Gammagard S/D:
1 Gm/kg. Up to 3 doses can be given on alternate days based on clinical response and platelet count.
Gammaked and gamunex-C:
1 Gm/kg/day (10 mL/kg/day) for 2 consecutive days for a total dose of 2 Gm/kg. May withhold the second 1 Gm/kg dose if an adequate platelet response is seen within 24 hours. Alternately, the 2 Gm/kg total dose can be administered as 400 mg/kg/day (4 mL/kg/day) for 5 consecutive days. The high-dose regimen (1 Gm/kg/day for 1 to 2 days) is not recommended for individuals with expanded fluid volumes or when fluid volume may be a concern.
Gammaplex, octagam 10%, and privigen:
Chronic ITP:
1 Gm/kg/day for 2 consecutive days for a total dose of 2 Gm/kg.
B-cell chronic lymphocytic leukemia (CLL)
Gammagard S/D:
400 mg/kg every 3 to 4 weeks.
Chronic inflammatory demyelinating polyneuropathy (CIDP)
Gamunex-C and gammaked:
Loading dose:
1 Gm/kg/day (10 mL/kg/day) for 2 consecutive days or 0.5 Gm/kg/day (5 mL/kg/day) for 4 consecutive days. With either regimen the total dose is 2 Gm/kg. Follow with maintenance dose of 1 Gm/kg every 3 weeks. The maintenance dose may be given as a total dose of 1 Gm/kg (10 mL/kg) on Day 1 or divided into 2 doses of 500 mg/kg/day given over 2 consecutive days.
Multifocal motor neuropathy (MMN)
Gammagard liquid:
Dose range is 0.5 to 2.4 Gm/kg/month based on clinical response.
Pediatric dose
Kawasaki syndrome
Gammagard S/D:
A single dose of 1 Gm/kg as an IV infusion. Alternately, 400 mg/kg/day may be administered for 4 consecutive days. Begin within 7 days of onset of fever. Concomitant administration of aspirin 80 to 100 mg/kg/day in 4 divided doses is indicated.
Other indications
See specific product manufacturer prescribing information for age restrictions for other indications.
See mg/kg or Gm/kg dose recommendations under Usual Dose. Begin with the lowest recommended dose. With most brands, clinical studies suggested that no difference in dosing is necessary and no special precautions are indicated. According to the manufacturers, infants and neonates were not included in clinical studies for primary immunodeficiency diseases or Kawasaki disease. All age-groups were represented in clinical studies for idiopathic thrombocytopenic purpura. See Maternal/Child.
Dose adjustments
Adjust dose according to IgG levels and clinical response. Frequency and dose will vary from patient to patient. No controlled clinical trials are available to determine optimum trough serum IgG level.
Dilution
Requirements for dilution of liquid and lyophilized powder preparations are brand specific and vary considerably; see manufacturer’s package insert. Some may be further diluted only with D5W or with D5W and/or NS; others cannot be diluted. Some require D5W or NS to flush the infusion line.
All formulations:
Do not mix with IGIV products from other manufacturers. Do not mix with other medicinal products. Administer through a separate infusion line or flush line before and after administration. Do not use if solution is cloudy, turbid, or contains particulate matter. Do not freeze. Do not use if previously frozen. Do not use beyond expiration date. For single use only; use promptly or discard unused product. Bring to room temperature before reconstitution (if required) or administration.
Most formulations:
Do not shake. If large doses are to be administered, several vials may be pooled using aseptic technique (time limits or specific processes may apply); see specific product.
Bivigam, Flebogamma, Gamunex-C, Gammagard Liquid, Gammaked, Gammaplex, Octagam, and Privigen are liquid preparations and are ready to use.
Carimune NF is a lyophilized product, and Gammagard S/D is a freeze-dried product. Both require reconstitution. Absolute sterile technique is required at all steps of the reconstitution process. Filtration may be required as drawn into a syringe for administration or as administered through IV tubing. Check both brands for specific equipment and specific dilution requirements.
Bivigam:
A ready-to-use 10% sterile solution; do not dilute. Available as 5 Gm in 50 mL or 10 Gm in 100 mL. Does not contain sucrose. Warm to room temperature before use and maintain at room temperature during administration. Do not shake. Several vials may be pooled using aseptic technique into sterile infusion bags and infused. pH 4 to 4.6.
Carimune NF:
Contains sucrose; see Precautions. Available as a lyophilized powder in 3-, 6-, and 12-Gm vials. Reconstitute with NS, D5W, or SWFI. Package insert has a chart with dilution requirements for all concentrations. Do not shake. Immediate use is recommended unless reconstituted under a sterile laminar flow hood; then it may be refrigerated and administration must begin within 24 hours. Several reconstituted vials of identical concentration and diluent may be pooled in an empty sterile glass or plastic IV infusion container using aseptic technique. pH 6.4 to 6.8. A 6% solution of Carimune NF is recommended for use in ITP.
Flebogamma:
Contains sorbitol; does not contain sucrose. A ready-to-use 5% (50 mg/mL)sterile solution; available as 0.5 Gm in 10 mL, 2.5 Gm in 50 mL, 5 Gm in 100 mL, 10 Gm in 200 mL, and 20 Gm in 400 mL. Also available as a ready-to-use 10% (100 mg/mL) sterile solution of 5 Gm in 50 mL, 10 Gm in 100 mL, and 20 Gm in 200 mL concentrations. Further dilution with other solutions not recommended. Using aseptic technique, several vials may be pooled into an empty sterile solution container. See Filters. pH 5 to 6.
Gammagard liquid:
Does not contain sucrose. A ready-to-use 10% solution available as 1 Gm in 10 mL, 2.5 Gm in 25 mL, 5 Gm in 50 mL, 10 Gm in 100 mL, and 20 Gm in 200 mL. Warm to room temperature before use. Do not shake. May be further diluted only with D5W. See Filters. pH 4.6 to 5.1.
Gammagard S/D:
Does not contain sucrose. A freeze-dried preparation. Must be warmed to room temperature before reconstitution if refrigerated. Diluent (SWFI), transfer device, administration set with integral airway, and filter provided with each single-use vial. Available in 2.5-, 5-, and 10-Gm single-dose vials with diluent. Use full amount of diluent (50, 96, 192 mL) to prepare a 5% solution (50 mg/mL), or remove one half the amount of diluent (25, 48, 96 mL) to prepare a 10% solution (100 mg/mL). Do not shake. Must be used within 2 hours of dilution if prepared outside a laminar flow hood. Administer within 24 hours if prepared aseptically inside a sterile laminar flow hood and stored in the original glass container or pooled into ViaFlex bags under constant refrigeration. Record date and time of reconstitution/pooling. pH 6.4 to 7.2.
Gammaked:
Does not contain sucrose. A ready-to-use 10% solution. Available as 1 Gm in 10 mL, 2.5 Gm in 25 mL, 5 Gm in 50 mL, 10 Gm in 100 mL, and 20 Gm in 200 mL. Bring to room temperature before administration if refrigerated. Use an 18-gauge needle or dispensing pin to penetrate the stopper of the 10-mL vial. Use 16-gauge needles or dispensing pins for the 25 mL and larger sizes. May be further diluted with D5W if required. Do not use any other diluent. Do not shake. Content of vials may be pooled under aseptic conditions into sterile infusion bags but must be infused within 8 hours of pooling. pH is 4 to 4.5.
Gammaplex:
Does not contain sucrose but does contain fructose; see Contraindications. A ready-to-use 5% sterile solution. Available as 2.5 Gm in 50 mL, 5 Gm in 100 mL, and 10 Gm in 200 mL. Do not shake. Begin infusion within 2 hours if multiple vials are pooled under aseptic conditions. Bring to room temperature before administration if refrigerated. pH 4.8 to 5.1.
Gamunex-C:
Does not contain sucrose. A ready-to-use 10% sterile solution. Available in 1-, 2.5-, 5-, 10-, and 20-Gm single-dose vials. Bring to room temperature before administration if refrigerated. Use an 18-gauge needle to penetrate the stopper of the 1-Gm size. Use a 16-gauge needle to penetrate the stopper of the 2.5- to 20-Gm sizes. Penetration of the stopper in the center of the raised ring and perpendicular to it is recommended. May be further diluted with D5W if required. Do not use any other diluent. Filtration during administration is not required. Use within 8 hours if multiple vials are pooled under aseptic conditions. pH 4 to 4.5.
Octagam:
Does not contain sucrose but does contain maltose; see Contraindications. A ready-to-use 5% or 10% sterile solution. The 5% solution is available as 1-, 2.5-, 5-, 10-, and 25-Gm single-dose vials. The 10% solution is available as 2- 5-, 10-, and 20-Gm single-dose vials. Do not shake. Use within 8 hours if multiple vials are pooled under aseptic conditions. Bring to room temperature before administration if refrigerated. pH 5.1 to 6 (5%); pH 4.5 to 5 (10%).
Privigen:
Does not contain sucrose but does contain l-proline; see Contraindications. A ready-to-use 10% sterile solution. Available in 5-, 10-, 20-, and 40-Gm single-dose vials. Do not shake. May be further diluted with D5W if required. Do not use any other diluent. Filtration during administration is not required. Content of vials may be pooled under aseptic conditions into sterile infusion bags but infusion should begin within 8 hours of pooling; contains no preservatives. pH 4.6 to 5.
Filters:
Filter or filter needle is provided by most manufacturers. Carimune NF and Flebogamma may be filtered with a larger pore filter size but is not required by manufacturers (filters greater than or equal to 15 microns will be less likely to slow infusion [0.2-micron antibacterial filters may be used with Carimune NF, Flebogamma, and Octagam]). Carimune NF is nano-filtered. Filtration of Gamunex-C during administration is not required; however, use of a non–protein-binding, 0.22-micron filter is permissible. Use of an in-line filter is optional with Gammagard Liquid and Octagam.
Storage:
Storage requirements are brand specific and vary considerably from refrigeration only to continuous or partial storage at RT not exceeding 30° C (77° F); see manufacturer’s package insert. Do not use after expiration date. Discard partially used vials. Do not use if solution is turbid or has been frozen. Some specifically state, “Do not heat or microwave.”
Compatibility
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
Manufacturers recommend administration through a separate IV line without admixture with other drugs and state, “Do not combine one IGIV product with an IGIV product from another manufacturer.” Gammagard Liquid, Gammaked, Gamunex-C, and Privigen are compatible only with D5W and are incompatible with NS. Flebogamma and Octagam should not be further diluted with any solution.
Most preparations may be infused sequentially into a primary IV of D5W or NS, or the tubing may be flushed with D5W or NS before and after administration.
One source suggests the following compatibilities: Not recommended by manufacturers.
Y-site:
Fluconazole (Diflucan), sargramostim (Leukine).
Rate of administration
All formulations:
Too-rapid infusion may cause a precipitous hypotensive reaction. Decrease rate of infusion at onset of patient discomfort or any adverse reactions; see Antidote. Decrease rate in patients at risk for neuromuscular disorders. See Precautions and individual products for suggested decrease in rates for patients at risk for thrombosis or developing renal dysfunction. Administer via separate IV tubing with filter or filter needle (provided by most manufacturers if required). Do not mix with IGIV products from other manufacturers or with other drugs or IV solutions. An infusion pump will facilitate an accurate rate of administration.
Bivigam:
Does not contain sucrose. 0.5 mg/kg/min for the first 10 minutes. If no discomfort or adverse effects, may be increased every 20 minutes by 0.8 mg/kg/min up to 6 mg/kg/min. In patients at risk for thrombosis or developing renal dysfunction, administer at the minimum dose and rate of infusion that is practicable.
Carimune NF:
Contains sucrose; see Precautions. An initial infusion rate of 0.5 mg/kg/min for 30 minutes. May then be increased to 1 mg/kg/min for the next 30 minutes. May be gradually increased in steps up to a maximum of 3 mg/kg/min as tolerated. The first dose in previously untreated agammaglobulinemic or hypogammaglobulinemic patients must be a 3% solution. After the first dose, subsequent infusions may be administered at a higher concentration as tolerated; see Precautions. In patients at risk for thrombosis or developing renal dysfunction, administer at the minimum dose; the maximum rate of infusion should not exceed 2 mg/kg/min.
Flebogamma:
Does not contain sucrose. In patients at risk for thrombosis or developing renal dysfunction, administer at the minimum dose and rate of infusion that is practicable.
Primary immunodeficiency (PI):
Initiate 5% solution at 0.01 mL/kg/min (0.5 mg/kg/min) for the first 30 minutes. If no discomfort or adverse effects, may be gradually increased to a maximum rate of 0.1 mL/kg/min (5 mg/kg/min).
Primary immunodeficiency (PI) and idiopathic thrombocytopenic purpura (ITP):
Initiate 10% solution at 0.01 mL/kg/min (1 mg/kg/min) for the first 30 minutes. If no discomfort or adverse effects, may be gradually increased to a maximum rate of 0.08 mL/kg/min (8 mg/kg/min).
Gammagard liquid:
Does not contain sucrose. In patients at risk for developing renal dysfunction or thrombotic episodes, the maximum infusion rate should be less than 3.3 mg/kg/min (less than 2 mL/kg/hr).
Primary immunodeficiency (PI):
0.5 mL/kg/hr (0.8 mg/kg/min) for the first 30 minutes. If no discomfort or adverse effects, may be gradually increased every 30 minutes to a maximum rate of 5 mL/kg/hr (8 mg/kg/min).
Multifocal motor neuropathy (MMN):
0.5 mL/kg/hr. (0.8 mg/kg/min) for the first 30 minutes. If no discomfort or adverse effects, may be gradually increased every 30 minutes to a maximum rate of 5.4 mL/kg/hr (9 mg/kg/min).
Gammagard S/D:
Dose not contain sucrose. 5% solution 0.5 mL/kg/hr. May be gradually increased to a maximum rate of 4 mL/kg/hr if no discomfort or adverse effects. If 5% solution well tolerated at 4 mL/kg/hr, a 10% solution can be used. Begin with 0.5 mL/kg/hr. If no adverse effects, gradually increase up to a maximum of 8 mL/kg/hr. In patients at risk for developing renal dysfunction, the concentration and infusion rate should be less than 3.3 mg/kg/min (less than 2 mL/kg/hr of 10% or less than 4 mL/kg/hr of 5%).
Gammaked:
Does not contain sucrose. In patients at risk for thrombosis or developing renal dysfunction, administer at the minimum dose and rate of infusion that is practicable.
Primary immunodeficiency (PI) and idiopathic thrombocytopenic purpura (ITP):
1 mg/kg/min (0.01 mL/kg/min). If the infusion is well tolerated, the rate may be gradually increased to a maximum of 8 mg/kg/min (0.08 mL/kg/min).
Chronic inflammatory demyelinating polyneuropathy (CDIP):
2 mg/kg/min (0.02 mL/kg/min). If the infusion is well tolerated, the rate may be gradually increased to a maximum of 8 mg/kg/min (0.08 mL/kg/min).
Gammaplex:
Does not contain sucrose. 0.5 mg/kg/min (0.01 mL/kg/min) for the first 15 minutes. If no discomfort or adverse effects, may be gradually increased every 15 minutes to 4 mg/kg/min (0.08 mL/kg/min). In patients at risk for thrombosis or developing renal dysfunction, administer at the minimum dose and rate of infusion that is practicable.
Gamunex-C:
Does not contain sucrose. In patients at risk for thrombosis or developing renal dysfunction, administer at the minimum dose and rate of infusion that is practicable.
Primary immunodeficiency (PI) and idiopathic thrombocytopenic purpura (ITP):
1 mg/kg/min (0.01 mL/kg/min) for the first 30 minutes. If no discomfort or adverse effects, may be gradually increased to a maximum rate of 8 mg/kg/min (0.08 mL/kg/min).
Chronic inflammatory demyelinating polyneuropathy (CIDP):
2 mg/kg/min (0.02 mL/kg/min). If no discomfort or adverse effects, may be gradually increased to a maximum rate of 8 mg/kg/min (0.08 mL/kg/min).
Octagam:
Does not contain sucrose; does contain maltose. In patients at risk for thrombosis or developing renal dysfunction, administer at the minimum dose and rate of infusion that is practicable.
Primary immunodeficiency (PI):
Initiate 5% solution at 0.5 mg/kg/min for the first 30 minutes. If no discomfort or adverse effects, may increase gradually in 30-minute increments to a maximum rate of 3.33 mg/kg/min.
Idiopathic thrombocytopenic purpura (ITP):
Initiate 10% solution at 1 mg/kg/min for the first 30 minutes. If no discomfort or adverse effects, may increase gradually in 30-minute increments to a maximum rate of 12 mg/kg/min.
Privigen:
Does not contain sucrose. IV line may be flushed with D5W or NS. In patients at risk for thrombosis or developing renal dysfunction, administer at the minimum dose and rate of infusion that is practicable.
Primary immunodeficiency:
0.5 mg/kg/min (0.005 mL/kg/min) initially. If no discomfort or adverse effects, may be gradually increased to a maximum rate of 8 mg/kg/min (0.08 mL/kg/min).
Chronic immune thrombocytopenic purpura:
0.005 mL/kg/min (0.5 mg/kg/min) initially. If no discomfort or adverse effects, may be gradually increased to a maximum rate of 0.04 mL/kg/min (4 mg/kg/min).
Actions
A preparation of concentrated human immunoglobulin G (IgG) antibodies. Obtained, purified, and standardized from human plasma. Specific methods during the manufacturing process (e.g., cold ethanol fractionation, detergents, solvents, nanofiltration) inactivate blood-borne viruses (e.g., hepatitis, HIV). Used as replacement therapy in primary and secondary immunodeficiencies. Active against bacterial, viral, parasitic, and mycoplasma antigens. Capable of both opsonization and neutralization of microbes and toxins.Also contains antibodies capable of interacting with and altering the activity of the immune system as well as antibodies capable of reacting with cells such as erythrocytes. (The role of these antibodies and the mechanisms of action of IgG have not been fully clarified.) Provides immediate antibody levels following infusion. The immediate peak in serum IgG is followed by a rapid decay due to equilibration between the plasma and extravascular fluid compartments. Half-life is variable (approximately 3 to 6 weeks) but may be decreased by fever or infection (increased catabolism or consumption). Crosses the placenta.
Indications and uses
Selected products are approved for different uses; see Usual Dose. All provide rapid-onset, short-term passive immunization. Labeled uses include: Primary immunodeficiency diseases: Replacement therapy in adults, adolescents, and other pediatric patients unable to produce adequate amounts of IgG antibodies, especially in the following situations: need for immediate increase in intravascular immunoglobulin levels, small muscle mass or bleeding tendencies that contraindicate IM injection, and selected disease states (e.g., congenital agammaglobulinemia, common variable immunodeficiency [CVID], combined immunodeficiency, X-linked agammaglobulinemia, Wiskott-Aldrich syndrome). ■ Treatment of acute and chronic idiopathic thrombocytopenic purpura (also called immune thrombocytopenic purpura or primary immune thrombocytopenia): Temporary increase in platelet counts in patients with idiopathic thrombocytopenic purpura and with thrombocytopenia associated with bone marrow transplant. ■ Treatment of chronic inflammatory demyelinating polyneuropathy (CIDP): Improvement of neuromuscular disability and impairment and maintenance therapy to prevent relapse. ■ Adjunct in chronic lymphocytic leukemia: Prevention of bacterial infections in patients with hypogammaglobulinemia or recurrent bacterial infection associated with B-cell CLL. ■ Multifocal motor neuropathy (MMN): To improve muscle strength and disability in adult patients. ■ Kawasaki syndrome: Prevention of coronary artery aneurysms associated with Kawasaki syndrome. ■ Safety and effectiveness of Gammagard S/D, Gammaplex, and Octagam 10% in pediatric patients with chronic ITP have not been established; see Maternal/Child for additional limitations with other products. ■ Several formulations are approved for SC use in specific diagnoses; consult prescribing information.
Unlabeled uses:
There are numerous unlabeled uses; consult literature.
Contraindications
Individuals known to have anaphylactic or severe hypersensitivity responses to the administration of human immune globulin. ■ IgA-deficient patients with pre-existing anti-IgA antibodies and a history of hypersensitivity. ■ Gammaplex is contraindicated in patients with hereditary intolerance to fructose; also contraindicated in infants and neonates for whom sucrose or fructose tolerance has not been established. ■ Octagam is contraindicated in patients with acute hypersensitivity to corn (contains maltose, which is derived from corn). ■ Privigen is contraindicated in patients with hyperprolinemia because it contains l-proline as a stabilizer.
Precautions
Check label; must state, “For IV use.” ■ Some formulations may be given SC; however, all products must be given by IV infusion for ITP patients. Do not administer SC due to a potential risk for hematoma. ■ Hypersensitivity reactions have occurred; administer in a facility with adequate equipment and supplies to monitor the patient and respond to any medical emergency. ■ Use extreme caution in individuals with a history of prior systemic hypersensitivity reactions. Incidence of anaphylaxis may be increased, especially with repeated injections. IgA-deficient patients, especially those with antibodies against IgA, are at greater risk for developing severe hypersensitivity reactions. ■ Some packaging of these products may contain latex; use caution in sensitive individuals. ■ IGIV products have been associated with renal dysfunction, acute renal failure, osmotic nephrosis, and death. Use extreme caution in patients with any degree of renal insufficiency; in patients age 65 years and older; in patients with diabetes mellitus, paraproteinemia, sepsis, or volume depletion; and/or in patients receiving known nephrotoxic drugs. If used, should be administered at the minimum dose and rate of infusion practicable. Products containing sucrose as a stabilizer (e.g., Carimune NF) have demonstrated an increased risk for renal dysfunction. Consider benefit versus risk before use. ■ Increases in SCr and/or BUN may progress to oliguria and anuria requiring dialysis; however, some patients improve spontaneously with discontinuation of IGIV. ■ May cause aseptic meningitis syndrome (AMS), especially with high doses (greater than 1 Gm/kg) and/or rapid infusion. May begin from 2 hours to 2 days after treatment. Symptoms are drowsiness, fever, headache (severe), nausea and vomiting, nuchal rigidity, painful eye movements, and photophobia. Cerebrospinal fluid (CSF) studies are often positive with pleocytosis, predominantly from the granulocyte series and elevated protein levels. ■ Hyperproteinemia, with resultant changes in serum viscosity, and electrolyte imbalances (e.g., hyponatremia) may occur. Distinguish true hyponatremia from pseudohyponatremia to determine correct treatment. Decreasing serum-free water in patients with pseudohyponatremia may lead to volume depletion with a further increase in serum viscosity, which may predispose to thromboembolic events. ■ IGIV products have been associated with thrombotic events. Risk factors may include advanced age, prolonged immobilization, hypercoagulable conditions, history of venous or arterial thrombosis, use of estrogens, indwelling vascular catheter, hyperviscosity, and cardiovascular risk factors (e.g., cerebrovascular disease, coronary artery disease, diabetes, hypertension). Thrombosis may occur in the absence of known risk factors. If used, should be administered at the minimum dose and rate of infusion practicable. Ensure adequate hydration before administration. ■ Baseline assessment of blood viscosity should be made in patients at risk for hyperviscosity, including those with cryoglobins, fasting chylomicronemia, markedly high triglycerides, or monoclonal gammopathies. ■ May rarely cause hemolysis, which can result in hemolytic anemia due to enhanced RBC sequestration. High doses (greater than 2 Gm/kg) and non-O blood group may be risk factors for hemolysis. Cases of severe hemolysis-related renal dysfunction/failure or DIC have occurred following infusion; see Monitor. ■ Consider risk/benefit before use of high-dose regimens (greater than 1 Gm/kg) in chronic ITP patients at increased risk for acute kidney injury, hemolysis, thrombosis, or volume overload. ■ Noncardiogenic pulmonary edema (transfusion-related acute lung injury [TRALI]) has been reported; see Monitor. ■ Derived from human blood. Despite purification processes, may carry risk of transmitting infectious agents (e.g., viruses [e.g., HIV, hepatitis] or Creutzfeldt-Jakob disease [CJD] agent). ■ Patients with gammaglobulinemia or extreme hypogammaglobulinemia who have never before received immunoglobulin substitution treatment or patients whose time from the last treatment is greater than 8 weeks may be at risk for developing inflammatory reactions (e.g., chills, fever, nausea, vomiting) on rapid infusion. Initiate slowly and increase rate as tolerated.
Monitor:
Use of larger veins recommended to reduce infusion-site discomfort, especially with 10% solutions. ■ Correct volume depletion before administration in all patients, especially patients with pre-existing renal insufficiency. ■ Recording of lot number on vials is recommended. ■ Monitor vital signs and observe patient continuously during infusion. A precipitous drop in BP or anaphylaxis can occur at any time. Emergency equipment and supplies must be at bedside. ■ Monitor renal function (e.g., BUN, SCr) and urine output in patients at increased risk for renal failure. Obtain baseline studies, monitor at intervals, and discontinue IGIV if renal function deteriorates. See Precautions. ■ Monitor for hyperproteinemia with resultant changes in serum viscosity and electrolyte imbalances. ■ Monitor for S/S of thrombosis and assess blood viscosity in patients at risk for hyperviscosity. ■ Monitor for S/S of hemolysis (e.g., lysis of red blood cells, liberation of hemoglobin), significant drop in hemoglobin or hematocrit, and hemolytic anemia. ■ Monitor for S/S of TRALI (e.g., fever, hypoxemia, normal left ventricular function, pulmonary edema, severe respiratory distress). Usually occurs within 1 to 6 hours after completion of the infusion. Manage with oxygen and adequate ventilatory support. If TRALI is suspected, both the product and the patient serum should be tested for the presence of antineutrophil antibodies. ■ Monitor for volume overload.
Patient education:
Report a burning sensation in the head, chills, cyanosis, diaphoresis, dyspnea, faintness or light-headedness, fatigue, fever, hives, itching or rash, neck pain or difficulty moving neck, tachycardia, wheezing. ■ Report chest pain or tightness, pain and/or swelling of an arm or a leg with warmth over the affected area, difficulty passing urine, decreased urine output, fluid retention, edema, shortness of breath, or sudden weight gain. ■ Remote risk of viral or CJD infection; consider risk versus benefit of therapy. See Drug/Lab Interactions.
Maternal/child:
Category C: use with caution in pregnancy; no adverse effects documented, but adequate studies are not available. ■ Safety for use in breast-feeding not established. ■ Response in pediatric patients usually exceeds response in adults. ■ Safety and effectiveness of Gammagard S/D in pediatric patients with chronic ITP have not been established. ■ Safety and effectiveness of Gammagard S/D in Kawasaki disease have been established. ■ Use of Privigen to treat primary immunodeficiency in pediatric patients under 3 years of age or to treat chronic immune thrombocytopenic purpura in pediatric patients under 15 years of age not established. ■ Safety and effectiveness of Bivigam not established for use in pediatric patients under 6 years of age. ■ Safety and effectiveness of Flebogamma 5% and 10% and Gammaplex in pediatric patients under 2 years of age have not been established. ■ Safety and effectiveness of Octagam 10% in pediatric patients have not been established. ■ Safety and effectiveness of Gammagard Liquid for treatment of PI in pediatric patients under 2 years of age have not been established. ■ Safety and effectiveness for use in pediatric patients with MMN have not been established. ■ Safety and effectiveness of Gammaked and Gamunex-C for use in pediatric patients with CIDP have not been established.
Elderly:
Use with extreme caution. Incidence of renal insufficiency, thrombosis, and other side effects increased due to age, potential for decreased organ function, and pre-existing medical conditions. Do not exceed recommended dose and infuse at the minimum infusion rate practicable; see Precautions.
Drug/lab interactions
Do not administer live virus vaccines from 2 weeks before to at least 3 months after immune globulin IV. Passive transfer of antibodies may transiently interfere with the response to live virus vaccines, such as measles, mumps, rubella, and varicella; see prescribing information if there is a risk of measles exposure or if accidental exposure has occurred. ■ Provides immediate antibody levels that last for about 3 weeks. In selected patients, may have an immune-modulating effect that may alter their response to corticosteroids or antineoplastic agents. ■ Concurrent use with nephrotoxic drugs (e.g., aminoglycosides [e.g., gentamicin], amphotericin B [Amphotec, conventional], cidofovir [Vistide], rifampin [Rifadin]) may increase risk of renal insufficiency. ■ Products that contain maltose (Octagam) may interfere with blood and urine glucose tests. ■ Various antibody titers may be raised temporarily, resulting in false-positive serologic testing. ■ May cause a positive direct or indirect antiglobulin (Coombs’) test.
Side effects
Arthralgia, asthenia, back pain, chills, diarrhea, dizziness, fatigue, fever, flushing, headache, hyperhidrosis, hypertension, infusion site pain/reactions, lethargy, nausea and vomiting, pharyngitis, rash, sinusitis, upper abdominal pain, and urticaria were reported most commonly. Full range of hypersensitivity symptoms, including anaphylaxis, is possible. Angioedema, erythema, fever, and urticaria are most frequently observed. Anxiety, chest tightness, cough, decreased diastolic BP, difficulty breathing, elevated ALT and AST (temporary), hemolytic anemia (reversible), increased BUN and SCr (may occur as soon as 1 to 2 days following infusion), leg cramps, light-headedness, malaise, migraine, myalgia, pharyngolaryngeal pain, and tachycardia have been reported. Severe reactions (e.g., circulatory collapse, fever, loss of consciousness, nausea and vomiting, sudden onset of dyspnea) have occurred and are more common in patients with antibody deficiencies. Noncardiogenic pulmonary edema (transfusion-related acute lung injury [TRALI]) has been reported. Acute renal failure, acute tubular necrosis, osmotic nephrosis, and proximal tubular nephropathy have been reported; may result in death. Is made from human plasma; process attempts to eliminate risk of hepatitis or HIV infection. A precipitous hypotensive reaction can occur and is most frequently associated with too-rapid rate of injection. Aseptic meningitis syndrome and hemolysis occur infrequently. See Precautions.
Post-marketing:
Abdominal pain, apnea, ARDS, back pain, bronchospasm, bullous dermatitis, cardiac arrest, coma, cyanosis, dyspnea, epidermolysis, erythema multiforme, hepatic dysfunction, hypotension, hypoxemia, leukopenia, loss of consciousness, pancytopenia, positive direct antiglobulin (Coombs’) test, pulmonary edema, seizures, Stevens-Johnson syndrome, thromboembolism, and tremor have been reported with IGIV products.
Antidote
Reduce rate for patient discomfort, for any sign of adverse reaction, and in patients at risk for renal insufficiency or thrombosis. If symptoms subside promptly, the infusion may be resumed at a lower rate. Decreasing the volume of subsequent infusions may also prevent or decrease the incidence of adverse reactions. Loop diuretics (e.g., furosemide [Lasix]) may be helpful in the management of fluid overload. Patients who continue to experience adverse reactions after rate and/or volume have been reduced may be premedicated with hydrocortisone 1 to 2 mg/kg 30 minutes before the immune globulin infusion. Pretreatment with acetaminophen (Tylenol) and diphenhydramine (Benadryl) or trying a different brand of immune globulin may also be useful. Discontinue the drug immediately for any signs of a hypersensitivity reaction, thrombotic event, or renal insufficiency. Notify the physician. May be treated symptomatically and infusion resumed at slower rate if symptoms subside. Treat anaphylaxis immediately. Epinephrine (Adrenalin), diphenhydramine (Benadryl), oxygen, vasopressors (e.g., dopamine), corticosteroids, and ventilation equipment must always be available. Manage TRALI with oxygen and ventilatory support. Resuscitate as necessary.
Indomethacin sodium
(in-doh-METH-ah-sin SO-dee-um)
Indocin IV
Prostaglandin inhibitor (patent ductus arteriosus adjunct)
pH 6 to 7.5
Usual dose
Neonates:
Three IV doses, specific to age at first dose, given at 12- to 24-hour intervals constitute a course of therapy.
Less than 48 hours of age:
First dose (0.2 mg/kg of body weight), second dose (0.1 mg/kg), third dose (0.1 mg/kg).
2 to 7 days of age:
0.2 mg/kg for each of 3 doses.
Over 7 days of age:
First dose (0.2 mg/kg), then 0.25 mg/kg for the next 2 doses.
If ductus arteriosus reopens, a second course of 1 to 3 doses as described for each neonate age may be repeated one time given at 12- to 24-hour intervals. If neonate remains unresponsive to indomethacin therapy after 2 courses, surgery may be required for closure of the ductus arteriosus.
Dose adjustments
If urine output is less than 0.6 mL/kg/hr at any time a dose is to be given, withhold dose until lab studies confirm normal renal function.
Dilution
Each 1 mg must be diluted with at least 1 mL NS or SWFI without preservatives (0.1 mg/0.1 mL); may be diluted with 2 mL diluent (0.05 mg/0.1 mL). The preservative benzyl alcohol is toxic in neonates. A fresh solution should be prepared just before each administration. Discard any unused portion.
Filters:
No data available from manufacturer.
Storage:
Store unopened vials in carton at CRT. Protect from light. Use reconstituted solution immediately.
Compatibility
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
Manufacturer states, “Prepare only with preservative-free NS or SWFI”; further dilution with IV solutions is not recommended and may precipitate with solutions with a pH below 6.
One source suggests the following compatibilities:
Y-site:
Dextrose 2.5% and 5%, furosemide (Lasix), insulin (regular), potassium chloride (KCl), sodium bicarbonate, nitroprusside sodium.
Rate of administration
A single dose, properly diluted, by IV injection over 20 to 30 minutes.
Actions
A potent inhibitor of prostaglandin synthesis. Through an unconfirmed method of action (thought to be inhibition of prostaglandin synthesis), it causes closure of a patent ductus arteriosus 75% to 80% of the time, eliminating the need for surgical intervention. Plasma half-life varies inversely with postnatal age and weight and ranges from 12 to 20 hours. Metabolized in the liver and eventually excreted in urine and bile.
Indications and uses
Closure of a hemodynamically significant patent ductus arteriosus in premature infants weighing between 500 and 1,750 Gm if usual medical management (e.g., fluid restriction, diuretics, digoxin, respiratory support) has not been effective after 48 hours.
Contraindications
Bleeding, especially active intracranial hemorrhage or GI bleeding; coagulation defects; necrotizing enterocolitis; infants with congenital heart disease (e.g., pulmonary atresia, severe coarctation of the aorta, severe tetralogy of Fallot) who require patency of the ductus arteriosus for satisfactory pulmonary or systemic blood flow; proven or suspected untreated infection; significant renal impairment; thrombocytopenia.
Precautions
Clinical evidence of a hemodynamically significant patent ductus arteriosus (respiratory distress, a continuous murmur, a hyperactive precordium, cardiomegaly and pulmonary plethora on chest x-ray) should be present before use is considered. ■ For use only in a highly supervised setting such as an intensive care nursery. ■ May increase potential for GI or intraventricular bleeding. ■ Use caution in presence of existing controlled infection; may mask signs and symptoms of exacerbation. ■ May suppress water excretion to a greater extent than sodium excretion. Hyponatremia may result. ■ For IV use only. ■ Surgery indicated if condition is not responsive to two courses of therapy.
Monitor:
Vital signs, oxygenation, acid-base status, fluid and electrolyte balance, and kidney function (SCr, BUN, urine output) must be monitored and maintained. ■ Can cause marked reduction in urine output (over 50%), increase BUN and SCr, and reduce glomerular filtration rate and CrCl. These symptoms usually disappear when therapy completed but may cause acute renal failure, especially in infants with impaired renal function from other causes. ■ May inhibit platelet aggregation; monitor for signs of bleeding. ■ Discontinue drug if signs of impaired liver function appear. ■ Confirm absolute patency of vein. Avoid extravasation; will irritate tissue. ■ See Drug/Lab Interactions.
Drug/lab interactions
May reduce elimination and increase serum concentrations of drugs that are renally excreted (e.g., aminoglycosides [e.g., gentamicin], digoxin [Lanoxin]). Monitor drug levels and adjust doses as needed to avoid toxicity. ■ Observe neonate closely for signs of digoxin toxicity; frequent monitoring of ECG and digoxin serum levels is indicated. ■ Use with furosemide (Lasix) may help to maintain renal function. ■ Concomitant use with anticoagulants may increase risk of bleeding; monitoring of PT suggested. ■ Coadministration with ACE inhibitors (e.g., enalapril [Vasotec]) may result in deterioration of renal function, including renal failure.
Side effects
Abdominal distension; acidosis; alkalosis; apnea; bleeding into the GI tract (gross or microscopic); bradycardia; DIC; elevated BUN or creatinine; exacerbation of preexisting pulmonary infection; fluid retention; gastric perforation; hyperkalemia; hypoglycemia; hyponatremia; intracranial bleeding; necrotizing enterocolitis; oliguria; oozing from needle puncture sites; pulmonary hemorrhage; pulmonary hypertension; reduced urine sodium, chloride, potassium, urine osmolality, free water clearance, or glomerular filtration rate; renal failure; retrolental fibroplasia; thrombocytopenia; transient ileus; uremia; vomiting.
Antidote
Discontinue the drug and notify the physician of all side effects. Based on severity, side effects may be treated symptomatically or drug will be completely discontinued in favor of surgical intervention. Resuscitate as necessary.
Infliximab  ■ infliximab-dyyb
■ infliximab-dyyb  *
*
(in-FLIX-ih-mab)
Remicade, Inflectra*
Monoclonal antibody
Inflammatory bowel disease agent
Antirheumatic agent
TNF-blocking agent
pH 7.2
Usual dose
Preliminary patient evaluation required; see Monitor.
Inflectra and remicade:
Premedication:
Administer at the physician’s discretion. May include antihistamines (e.g., diphenhydramine [Benadryl]), H2 blockers (e.g., famotidine [Pepcid IV]), acetaminophen, and/or corticosteroids (e.g., hydrocortisone).
Crohn’s disease and fistulizing Crohn’s disease:
Begin with an initial dose of 5 mg/kg as an infusion. Repeat at 2 and 6 weeks and every 8 weeks thereafter. For patients who respond and then lose their response, consideration may be given to treatment with 10 mg/kg. If there is no response by Week 14, response with continued dosing is unlikely; consider discontinuing infliximab. See Precautions.
Rheumatoid arthritis:
3 mg/kg as an infusion. Repeat dose at 2 and 6 weeks, then every 8 weeks thereafter. Given in combination with methotrexate at a minimum dose of 12.5 mg/week (median dose of 15 mg/week). See methotrexate monograph. If response to infliximab is incomplete, dose may be adjusted up to 10 mg/kg or interval decreased to every 4 weeks. Risk of infection may be increased at higher doses.
Ankylosing spondylitis:
5 mg/kg as an infusion. Repeat at 2 and 6 weeks, then every 6 weeks thereafter.
Psoriatic arthritis:
5 mg/kg as an infusion. Repeat dose at 2 and 6 weeks, then every 8 weeks thereafter. May be used with or without methotrexate.
Ulcerative colitis and plaque psoriasis:
5 mg/kg as an infusion. Repeat dose at 2 and 6 weeks, then every 8 weeks thereafter.
Pediatric dose
Preliminary patient evaluation required; see Monitor. See Maternal/Child.
Crohn’s disease (inflectra and remicade) and ulcerative colitis† (remicade):
5 mg/kg as an infusion. Repeat dose at 2 and 6 weeks. Follow with a maintenance regimen of 5 mg/kg every 8 weeks.
Dose adjustments
Inflectra and remicade:
Do not exceed a dose of 5 mg/kg in patients with moderate to severe CHF (NYHA Class III/IV); see Contraindications.
Dilution
Inflectra and remicade:
Each vial contains 100 mg of infliximab. When reconstituted as directed below, each milliliter of solution contains 10 mg of infliximab. Calculate the dose and the number of vials required and the total volume of reconstituted infliximab solution required. Reconstitute each 100-mg vial with 10 mL of SWFI, using a syringe equipped with a 21-gauge or smaller needle. Direct the stream of SWFI to side of vial. Swirl gently; do not shake. Allow reconstituted solution to stand for 5 minutes. Solution should be colorless to light yellow and opalescent and may develop a few translucent particles as infliximab is a protein. Do not use if opaque particles, discoloration, or other foreign particles are present. The total dose of reconstituted solution must be further diluted with NS to a final volume of 250 mL. (May withdraw a volume of NS equal to the calculated volume of reconstituted infliximab from a 250-mL bottle or bag of NS and slowly add reconstituted solution.) Do not dilute the reconstituted infliximab solution with any other diluent. Mix gently. Infusion concentration should range between 0.4 mg/mL and 4 mg/mL. The infusion should begin within 3 hours of reconstitution and dilution.
Filters:
Inflectra and remicade:
Must be administered through an infusion set with an in-line, sterile, nonpyrogenic, low–protein binding filter (pore size equal to or less than 1.2 microns). Flush and prime tubing/filter system with NS before administration of infusion.
Storage:
Inflectra and remicade:
Refrigerate unopened vials at 2° to 8° C (36° to 46° F). Discard any unused portion. Do not use beyond expiration date on vials.
Compatibility
Inflectra and remicade:
Manufacturer recommends not infusing concomitantly in the same IV line with other agents until specific compatibility data are available.
Rate of administration
Inflectra and remicade:
Begin the infusion within 3 hours of reconstitution and dilution. Flush and prime tubing/filter system with NS before administration. A single dose should be given over a period of not less than 2 hours. Upon completion of infusion, IV line should be flushed thoroughly with 15 to 20 mL of NS to ensure all active drug is delivered to the patient. Patients experiencing a mild to moderate infusion-related reaction may be able to continue therapy at a reduced rate; see Antidote.
Actions
Inflectra and remicade:
A chimeric IgG1 monoclonal antibody that binds specifically to human tumor necrosis factor alpha (TNFα). Is composed of human constant and murine variable regions. Neutralizes the biologic activity of TNFα by binding with high affinity to the soluble and transmembrane forms of TNFα and inhibiting binding of TNFα with its receptors. Infliximab does not neutralize TNFβ. Biologic activities attributed to TNFα include induction of pro-inflammatory cytokines such as IL-1 and IL-6, enhancement of leukocyte migration by increasing endothelial layer permeability and expression of adhesion molecules by endothelial cells and leukocytes, activation of neutrophil and eosinophil functional activity, and induction of acute phase reactants and other liver proteins, as well as tissue-degrading enzymes produced by synoviocytes and/or chondrocytes. Elevated concentrations of TNFα have been found in involved tissues and fluids of patients with Crohn’s disease, rheumatoid arthritis, ankylosing spondylitis, ulcerative colitis, psoriatic arthritis, and plaque psoriasis. These elevated concentrations correlate with elevated disease activity. Treatment with infliximab blocks the biological activities attributed to TNFα. After treatment, patients have decreased levels of serum IL-6 and C-reactive protein compared to baseline. Infliximab is distributed predominantly within the vascular space. Has a prolonged terminal half-life of 7.7 to 9.5 days. Produced by a recombinant cell line cultured by continuous perfusion and is purified by a series of steps that include measures to inactivate and remove viruses.
Indications and uses
Adult and pediatric patients 6 years of age and older:
Reduce the S/S and induce and maintain clinical remission in patients with moderately to severely active Crohn’s disease who have had an inadequate response to conventional therapy (Inflectra and Remicade). ■ Reduce the S/S and induce and maintain clinical remission and mucosal healing in patients with moderately to severely active ulcerative colitis who have had an inadequate response to conventional therapy. (Inflectra and Remicade may be used in adults; however, use in pediatric patients is limited to Remicade due to marketing exclusivity for Remicade.) In addition, Inflectra and Remicade are indicated to help eliminate corticosteroid use in adult patients with ulcerative colitis.
Adult patients:
Inflectra and remicade:
Reduce the number of draining enterocutaneous and rectovaginal fistula(s) and maintain fistula closure in fistulizing Crohn’s disease. ■ Given in combination with methotrexate to improve physical function, inhibit progression of structural damage, and reduce S/S in patients with moderately to severely active rheumatoid arthritis. ■ Reduce S/S in active ankylosing spondylitis. ■ Reduce the S/S of active arthritis, inhibit progression of structural damage, and improve physical function in psoriatic arthritis. ■ Used for treatment of patients with chronic severe (i.e., extensive and/or disabling) plaque psoriasis who are candidates for systemic treatment and when other systemic treatments are less appropriate.
Contraindications
Inflectra and remicade:
Known hypersensitivity to infliximab, murine proteins, or other components of the product. ■ Administration of doses exceeding 5 mg/kg in patients with moderate to severe (NYHA Class III/IV) CHF.
Precautions
Inflectra and remicade:
Administer in a facility that is equipped to monitor the patient and respond to any medical emergency. ■ TNFα mediates inflammation and modulates cellular immune response. Anti-TNF therapies, including infliximab, may affect normal immune responses. ■ Patients treated with infliximab are at increased risk for developing serious infections that may lead to hospitalization or death. Many of the serious infections have occurred in patients undergoing concomitant immunosuppressive therapy (e.g., methotrexate, corticosteroids) that, in addition to their underlying disease, may predispose them to infections; see Drug/Lab Interactions. Discontinue infliximab if patient experiences a serious infection or sepsis. Reported infections with TNF-blockers include active tuberculosis, including reactivation of latent tuberculosis; invasive fungal infections (e.g., histoplasmosis, coccidioidomycosis, candidiasis, aspergillosis, blastomycosis, and pneumocytosis); and bacterial, viral, and other infections due to opportunistic pathogens, including Legionella and Listeria. ■ Patients with TB have frequently presented with disseminated or extrapulmonary disease. ■ Cases of tuberculosis reactivation or new tuberculosis infections have been observed in patients receiving infliximab, including patients who have previously received treatment for latent or active tuberculosis. Cases of active tuberculosis have also occurred in patients being treated with infliximab during treatment for latent tuberculosis. ■ Antituberculosis treatment of patients with a reactive TB skin test reduces the risk of TB reactivation in patients receiving infliximab. ■ Patients with histoplasmosis or other invasive fungal infections may present with disseminated rather than localized disease. Antigen and antibody testing for histoplasmosis may be negative. ■ Treatment with infliximab should not be initiated in patients with an active infection, including clinically important localized infections. Consider the risks and benefits of infliximab treatment before initiating therapy in patients with chronic or recurrent infection, patients who have been exposed to TB, patients with a history of an opportunistic infection, patients who have resided in or traveled to areas of endemic tuberculosis or endemic mycoses (e.g., histoplasmosis, coccidioidomycosis, or blastomycosis), or patients with underlying conditions that may predispose them to infection. ■ Has been associated with the reactivation of hepatitis B virus (HBV) in patients who are chronic hepatitis B carriers (i.e., surface antigen–positive). Fatalities have been reported. ■ Infliximab therapy has been associated with adverse outcomes in patients with heart failure and should be used in these patients only after consideration of other treatment options. A higher incidence of worsening CHF, increased mortality, and a higher rate of cardiovascular adverse events have been seen in patients with moderate to severe CHF. Occurs more frequently in patients receiving a 10 mg/kg dose. Do not use doses greater than 5 mg/kg in patients with moderate to severe CHF; see Contraindications. Discontinue for new-onset CHF or for worsening CHF, and consider discontinuing in patients with CHF who have not had a significant response to infliximab. Use with caution in patients with mild CHF (NYHA Class I/II); monitor closely. ■ Hypersensitivity reactions characterized by urticaria, dyspnea, and/or hypotension have occurred in association with infliximab infusion. Most occur during or within 2 hours of the infusion. Discontinue infusion if severe reaction occurs. Medications for management of hypersensitivity reaction (e.g., acetaminophen [Tylenol], diphenhydramine [Benadryl], corticosteroids [e.g., hydrocortisone] and/or epinephrine) should be readily available. ■ Serum sickness–like reactions (dysphagia, fever, hand and facial edema, headache, myalgias, polyarthralgias, sore throat) have been reported and have occurred as early as after the second dose and when infliximab was interrupted and then re-initiated after an extended period. These reactions are associated with a marked increase in antibodies to infliximab, a loss of detectable serum concentrations of infliximab, and a possible loss of drug efficacy. ■ Readministration of infliximab after a period of no treatment resulted in a higher incidence of infusion reactions relative to regular maintenance treatment in clinical trials. Evaluate risk versus benefit of readministration after a period of no treatment, especially if considering a re-induction regimen given at 0, 2, and 6 weeks. For cases in which maintenance therapy for psoriasis has been interrupted, infliximab should be re-initiated with a single dose followed by maintenance therapy. ■ Infliximab therapy may result in formation of autoimmune antibodies and, rarely, in the development of a lupus-like syndrome. Discontinue therapy if this occurs. In clinical studies, symptoms resolved with discontinuation of therapy. ■ Lymphoma and other malignancies, some fatal, have been reported in children and adolescent patients treated with TNF blockers, including infliximab. Most of the affected patients were receiving concomitant immunosuppressants. ■ Cases of hepatosplenic T-cell lymphoma have been reported. The majority of reported cases have occurred in adolescents and young adult males being treated with infliximab for Crohn’s disease or ulcerative colitis. This type of lymphoma has a very aggressive disease course and is usually fatal. Almost all affected patients received concomitant treatment with a TNF-blocker and azathioprine or 6-mercaptopurine (Purinethol) at or before diagnosis. When treating patients with inflammatory bowel disease, particularly adolescents and young adults, the decision whether to use infliximab alone or in combination with other immunosuppressants should consider the possibility that there is a higher risk of developing hepatosplenic T-cell lymphoma (HSTCL) with combination therapy versus an observed increased risk of immunogenicity and hypersensitivity reactions with infliximab monotherapy. ■ Use caution in patients with moderate to severe COPD; may have an increased risk of malignancy, especially of the lungs and head or neck. ■ The potential role of TNF-blocking therapy in the development of lymphoma, leukemia, melanoma, Merkel cell carcinoma, and other malignancies is not known. Patients with Crohn’s disease, ulcerative colitis, rheumatoid arthritis, or plaque psoriasis, particularly with highly active disease and/or exposure to immunosuppressant therapies, may be at higher risk (up to several-fold) than the general population for the development of lymphoma, leukemia, and other malignancies, even in the absence of TNF-blocking therapy. Use caution when considering infliximab therapy in patients with a history of malignancy or when continuing treatment in patients who develop a malignancy while receiving infliximab. ■ In clinical trials, nonmelanoma skin cancers were more common in psoriasis patients with previous phototherapy. ■ Rare cases with CNS manifestations of systemic vasculitis, seizures, and new-onset or exacerbation of clinical symptoms and/or radiographic evidence of CNS demyelinating disorders (e.g., multiple sclerosis, optic neuritis) and peripheral demyelinating disorders (e.g., Guillain-Barré syndrome) have been reported. Use with caution in patients with any of these existing neurologic disorders. Consider discontinuing therapy if any of these disorders develop. ■ Severe hepatic reactions, including liver failure, jaundice, hepatitis, cholestasis, and autoimmune hepatitis, have been reported. Reactions have occurred anywhere from 2 weeks to more than a year after initiation of treatment and have resulted in death or the need for a liver transplant. ■ Leukopenia, neutropenia, thrombocytopenia, and pancytopenia, some with fatal outcome, have been reported. ■ Use caution when switching between biological disease-modifying antirheumatic drugs (e.g., abatacept [Orencia], adalimumab [Humira], anakinra [Kineret], etanercept [Enbrel]) or other TNF-blocking agents (e.g., golimumab [Simponi Aria]); overlapping biological activity may further increase the risk of infection; see Drug/Lab Interactions. ■ Administration of live virus vaccines or therapeutic infectious agents (e.g., BCG bladder instillation for treatment of cancer) concurrently with infliximab is not recommended. Could result in clinical infections, including disseminated infections. ■ See Drug/Lab Interactions and Maternal/Child.
Monitor:
Inflectra and remicade:
Evaluate patients for tuberculosis risk factors and test for latent infection before initiating infliximab and periodically during therapy. Initiate treatment of latent TB before therapy with infliximab. Antituberculosis therapy should also be considered before initiating infliximab in patients with a history of latent or active tuberculosis for whom an adequate course of treatment cannot be confirmed and in patients with a negative test for latent tuberculosis but risk factors for tuberculosis infection. Consultation with a physician with expertise in the treatment of TB is recommended to aid in deciding whether initiating antituberculosis therapy is appropriate for a given patient. ■ During and after treatment, monitor for the development of tuberculosis in patients who tested negative for latent tuberculosis infection before initiating therapy. Tests for latent tuberculosis infection may also be falsely negative while on therapy with infliximab. Tuberculosis should be strongly considered in patients who develop a new infection during infliximab therapy, especially in patients who have previously or recently traveled to countries with a high prevalence of TB or who have had close contact with a person with active TB. ■ Before initiating TNF-blocker therapy, including infliximab, patients should be tested for HBV infection. Consultation with a physician with expertise in the treatment of hepatitis B is recommended if a patient tests positive for hepatitis B surface antigen. ■ In patients who are carriers of HBV and require treatment with TNF-blockers, closely monitor for clinical and laboratory signs of active HBV infection throughout treatment and for several months after completion of treatment. If HBV reactivation occurs, discontinue infliximab; see Antidote. ■ Monitor for S/S of infection during and after treatment; discontinue if a serious infection occurs and initiate appropriate antimicrobial treatment. Empiric antifungal therapy may be appropriate pending results of a diagnostic workup in patients at risk for invasive fungal infections; see Precautions. ■ Monitor cardiac status closely for new-onset or worsening CHF; see Precautions. ■ Patients may develop antibodies to infliximab. In clinical trials, patients who were antibody-positive were more likely to have higher rates of clearance and reduced efficacy and to experience an infusion reaction than were patients who were antibody-negative. The incidence of antibody development was lower among patients receiving immunosuppressant therapies (e.g., 6-mercaptopurine, azathioprine, corticosteroids). ■ Monitor for S/S of hepatotoxicity (e.g., jaundice and/or marked liver enzyme elevations [equal to or greater than 5 times the upper limit of normal]). ■ Monitor CBC with differential and platelet count periodically. ■ Monitor for S/S of hypersensitivity or infusion reaction (e.g., anaphylaxis, chills, dyspnea, fever, flu-like symptoms, GI symptoms, headache, hypotension, rash). ■ Monitor BP and pulse every 30 minutes during infusion. If patient experiences a significant change in vital signs (e.g., drop in diastolic BP of 15 to 20 mm Hg) or exhibits any symptoms that may indicate hypersensitivity, stop infusion. Evaluate symptoms and treat appropriately. Continue to monitor patient for at least 30 minutes after completion of the infusion. ■ Monitor for S/S of malignancies such as HSTCL (e.g., abdominal pain, hepatomegaly, night sweats, persistent fever, weight loss) and lymphoma. ■ Periodic skin examination is recommended for all patients, particularly those with risk factors for skin cancer.
Patient education:
Inflectra and remicade:
Read manufacturer’s medication guide before each treatment with infliximab. ■ Tell health care professionals of heart conditions, previous or current infections, and recent or past exposure to TB or histoplasmosis. ■ Review all medicines and disease history with pharmacist or physician before initiating treatment. ■ Promptly report abdominal pain, fever, S/S of heart failure (e.g., shortness of breath, swelling feet), infection, numbness, tingling, or visual disturbances. ■ Report S/S of hypersensitivity reactions (e.g., itching, rash, swelling in the throat). Usually occur during or immediately following the infusion but may occur from 3 to 12 days later. ■ Report changes of growths on your skin during or after infliximab treatment. ■ Report symptoms of a cytopenia such as bruising, bleeding, or persistent fever. ■ Report recently receiving or being scheduled to receive a live virus vaccine or treatment with a weakened bacteria (such as BCG for bladder cancer) to your physician.
Maternal/child:
Inflectra and remicade:
Category B: use during pregnancy only if clearly needed. ■ Has potential for harm to the nursing infant; discontinue breast-feeding. ■ At least a 6-month waiting period following birth is recommended before the administration of any live vaccine to infants exposed in utero to infliximab. Infliximab crosses the placental barrier and has been detected in these infants up to 6 months after the last dose of infliximab to the mother, and these infants may be at increased risk for infection. ■ Safety and effectiveness for use in pediatric patients with ulcerative colitis and/or Crohn’s disease who are less than 6 years of age not established. ■ Safety and effectiveness for use in pediatric patients with juvenile rheumatoid arthritis was evaluated in a multicenter trial. The study failed to establish effectiveness; see prescribing information for details. ■ Before initiating infliximab therapy in pediatric patients with Crohn’s disease or ulcerative colitis, all vaccinations should be brought up-to-date. ■ Long-term (greater than 1 year) safety and effectiveness in pediatric patients with Crohn’s disease not established.
Elderly:
Inflectra and remicade:
Specific differences in safety and effectiveness not noted; the incidence of serious side effects may be increased. An increase in the incidence of serious infections has been noted in patients 65 years and older, and because there is a higher incidence of infections in the elderly population in general, caution should be used in treating the elderly. See Precautions.
Drug/lab interactions
Inflectra and remicade:
Specific interaction studies have not been performed. ■ Concurrent administration with anakinra (Kineret), an interleukin-1 antagonist, or abatacept (Orencia), an antirheumatic agent, may be associated with an increased risk of serious infections. The added benefit of combination therapy has not been documented. Anakinra is also associated with an increased risk of neutropenia. Concurrent administration of TNF α-blocking agents (e.g., infliximab) with anakinra or abatacept is not recommended. ■ May cause increased immunosuppression and increased risk of infection with tocilizumab (Actemra); concurrent use should be avoided. ■ Concurrent use of infliximab with other biological products (adalimumab [Humira], etanercept [Enbrel], golimumab [Simponi Aria]) used to treat the same conditions as infliximab is not recommended; may increase risk of infection. ■ The majority of patients with Crohn’s disease, rheumatoid arthritis, or psoriatic arthritis received one or more of the following concomitant medications without evidence of any type of negative drug interaction: aminosalicylates, antibiotics, antivirals, corticosteroids, 6-mercaptopurine (Purinethol), azathioprine, folic acid, methotrexate, narcotics, NSAIDs (e.g., ibuprofen [Motrin, Advil], naproxen [Aleve, Naprosyn]), and sulfasalazine. ■ Patients receiving immunosuppressants (e.g., 6-mercaptopurine, azathioprine, corticosteroids) tended to experience fewer infusion reactions as compared to patients on no immunosuppressants. ■ Concomitant methotrexate use may decrease incidence of anti-infliximab antibody production and increase infliximab concentrations. ■ Live virus vaccines and therapeutic infectious agents (e.g., BCG for bladder instillation for treatment of cancer) should not be given concurrently with infliximab. Could result in clinical infection, including disseminated infection. In addition, it is also recommended that live vaccines not be given to infants after in utero exposure to infliximab for at least 6 months following birth; see Precautions and Maternal/Child. ■ Cytochrome P450 enzymes may be suppressed by increased levels of cytokines during inflammation. Infliximab antagonizes cytokine activity, and the formation of the CYP450 enzymes may be normalized. When initiating or discontinuing infliximab in patients treated with CYP450 substrates with a narrow therapeutic index, monitoring of the effect (e.g., warfarin [Coumadin]) or drug concentrations (e.g., cyclosporine [Sandimmune] or theophylline) is recommended. Dose adjustment may be necessary.
Side effects
Inflectra and remicade:
The most commonly reported side effects include abdominal pain, headache, infection (e.g., pharyngitis, sinusitis, upper respiratory tract, and urinary tract), and infusion-related reactions. The most common reasons for discontinuation of therapy were infusion-related reactions (e.g., dyspnea, fever, flushing, headache, rash) occurring during or within 2 hours of infusion or infections (bacterial, fungal, protozoal, and viral). See Precautions. Other less common side effects include abscess, anaphylactic-like reactions (including laryngeal/pharyngeal edema and severe bronchospasm), anemia, anxiety, arthralgia, autoantibodies/lupus-like syndrome, back pain, conjunctivitis, constipation, depression, diarrhea, dizziness, dyspepsia, dysuria, flushing, hot flashes, HACA development, hepatitis B virus reactivation, hepatotoxicity (autoimmune hepatitis, increased liver function tests, jaundice, liver failure), insomnia, intestinal obstruction, lymphoproliferative disorders, malignancies including lymphoma, moniliasis, myalgia, pain, paresthesia, peripheral edema, rash, sarcoidosis, seizures, serum sickness–like reactions, stomatitis, tachycardia, vertigo, and visual disturbances. See Precautions. Infections (bacterial, fungal, protozoal, and viral) including TB, invasive opportunistic infections (e.g., histoplasmosis, listeriosis, and pneumocystosis), new-onset or worsening CHF, CNS demyelinating disorders, and deaths have been reported.
Post-marketing:
Inflectra and remicade:
Cholestasis, erythema multiforme, hepatitis, hepatosplenic T-cell lymphomas, idiopathic and/or thrombotic thrombocytopenic purpura, interstitial lung disease (including pulmonary fibrosis/interstitial pneumonitis and, very rarely, rapidly progressive disease), jaundice, liver failure, melanoma and Merkel cell carcinoma, myocardial ischemia/infarction, neuropathies, neutropenia, pericardial effusion, peripheral demyelinating disorders (e.g., Guillain-Barré syndrome, chronic inflammatory demyelinating polyneuropathy, and multifocal motor neuropathy), psoriasis (including new-onset and pustular, primarily palmar/plantar), Stevens-Johnson syndrome, systemic and cutaneous vasculitis, toxic epidermal necrolysis, and transverse myelitis have been reported; fatalities have occurred.
Pediatric patients with Crohn’s disease:
Inflectra and remicade:
Anemia, bacterial infection, blood in stool, bone fracture, flushing, leukopenia, neutropenia, respiratory tract allergic reactions, and viral infections were reported more commonly in pediatric patients than in adult patients receiving similar treatment regimens. Serious side effects were infections (some fatal), including opportunistic infections and tuberculosis, infusion reactions, and hypersensitivity reactions. Malignancies (including hepatosplenic T-cell lymphomas), transient hepatic enzyme abnormalities, lupus-like syndromes, and the development of autoantibodies were less common, yet serious, reactions.
Antidote
Inflectra and remicade:
Notify physician of any side effects; most will be treated symptomatically. Discontinue infliximab if patient experiences a serious infection, significant changes in vital signs, new or worsening S/S of heart failure, hepatotoxicity, a hypersensitivity reaction, a lupus-like syndrome (fever, pleuritic pain, pleural effusion), a neurologic disorder, or significant hematologic abnormalities. Treat hypersensitivity and/or infusion reactions with acetaminophen (Tylenol), antihistamines (diphenhydramine [Benadryl]), corticosteroids, dopamine, and epinephrine as indicated. Slow or suspend infusion for a mild or moderate infusion-related reaction. Upon resolution of the reaction, re-initiation at a lower infusion rate and/or therapeutic administration of antihistamines, acetaminophen, and/or corticosteroids may be attempted with caution. If patient does not tolerate the infusion after these interventions, infliximab should be discontinued; see Usual Dose. Lupus-like syndrome usually subsides within 10 days of discontinuing infliximab. Discontinue infliximab in patients who develop HBV reactivation, and initiate antiviral therapy with appropriate supportive treatment. Safety of resuming therapy after HBV is controlled is not known; use caution if resumption of therapy is considered, and monitor closely.
Insulin injection (regular) ■ insulin aspart rDNA origin ■ insulin glulisine rDNA origin ■ insulin lispro rDNA origin
(IN-sue-lin in-JEK-shun ■ IN-sue-lin AS-part)
Humulin R, Novolin ge Toronto  ■ NovoLog ■ Apidra ■ Humalog
■ NovoLog ■ Apidra ■ Humalog
Hormone
Antidiabetic agent
pH 7 to 7.8 ■ pH 7.2 to 7.6 ■ pH 7.3 ■ pH 7 to 7.8
Usual dose
Only regular insulin, insulin aspart, insulin glulisine, and insulin lispro may be given IV. Confirm by label or package insert that a particular insulin product is for IV use; see Precautions.
Dose varies greatly. Will be dependent on patient’s condition and response. Insulin requirements may be altered during stress or major illness or with changes in exercise, meal pattern, or coadministered drugs. It is imperative that dosing is individualized and adjusted based on blood glucose determinations because of the marked loss of insulin from adsorption to glass and plastic infusion containers and tubing; see Precautions.
Low-dose treatment in ketoacidosis:
The American Diabetes Association recommends 0.15 unit/kg as a loading dose (10.5 units for a 70-kg patient), followed by a continuous infusion of 0.1 unit/kg/hr (7 units/hr for a 70-kg patient). Plasma glucose should decrease at a rate of 50 to 75 mg/dL/hr. If plasma glucose concentrations do not fall by 50 mg/dL within the first hour of insulin therapy, the insulin infusion rate may be doubled every hour, provided the patient is adequately hydrated. Decrease the rate of infusion when plasma glucose reaches 250 to 300 mg/dL. Start a separate IV of D5/1/2NS when plasma glucose reaches 300 mg/dL. Administer insulin to maintain serum glucose concentrations between 150 and 200 mg/dL in patients with diabetic ketoacidosis. Continue until metabolic acidosis is corrected. Administer an appropriate dose of insulin SC 30 minutes before discontinuing the insulin infusion (intermediate-acting insulin recommended).
Diabetes mellitus with hyperosmolar coma, nonketotic (unlabeled):
An initial bolus dose of 0.1 unit/kg followed by a continuous infusion of 0.1 unit/kg/hr until the blood glucose falls to 250 mg/dL.
Hyperkalemia (unlabeled):
Insulin may be used in patients with or without diabetes mellitus to treat hyperkalemia. Must be administered with a dextrose solution in nondiabetic patients. Begin with an initial dose of 5 to 10 units of regular insulin and 50 mL of dextrose 50% as an IV bolus. Follow with a continuous infusion of 10% dextrose with regular insulin 20 units/L. Administer at 50 mL/hr to prevent fasting hyperkalemia.
Diagnosis of growth hormone deficiency (regular insulin [unlabeled use]):
0.05 to 0.15 unit/kg of body weight as a rapid, one-time injection.
Pediatric dose
Ketoacidosis or hyperosmolar hyperglycemia:
Fluids:
Consider initial potential for dehydration and administer 10 to 20 mL/kg of NS or LR over 1 hour.
Regular insulin and insulin aspart:
Administer 0.1 unit/kg/hr as a continuous infusion. Goal is to reduce glucose by 80 to 100 mg/dL/hr. When glucose reaches 250 to 300 mg/dL, or if glucose decreases more than 100 mg/dL/hr, add D5W to fluids. SC insulin may be started once pH and bicarbonate are within normal limits. Discontinue insulin infusion 1 hour after SC dose. Monitor glucose levels and serum electrolytes and continue fluid replacement as appropriate.
Dose adjustments
A reduced dose of insulin may be indicated when infusions are discontinued and SC administration is indicated. As in Usual Dose, base on blood glucose determinations and patient’s response to therapy. ■ Dose requirements may be reduced in patients with renal or hepatic impairment. ■ Dose adjustment may be indicated if it is necessary to change from one insulin product to another.
Dilution
Regular insulin:
Use only if clear. May be given undiluted either directly into the vein or through a Y-tube or three-way stopcock. Another regimen adds 100 units of insulin (1 mL) to 100 mL of NS. This solution yields 1 unit/mL and is usually given at a rate of 0.1 unit/kg of body weight/hr (70-kg adult would receive 7 units/hr).
Insulin aspart:
May be diluted in NS to a final concentration of 0.05 to 1 unit/mL. Use a polypropylene infusion bag. See Precautions.
Humulin R U-100:
Must be diluted with NS to achieve a final concentration of 0.1 to 1 unit/mL (100 units of insulin [1 mL] to 100 mL of NS yields 1 unit/mL). Use polyvinyl chloride (PVC) infusion bags.
Insulin glulisine:
May be diluted only with NS to achieve a final concentration of 0.05 unit/mL to 1 unit/mL (e.g., dilute 100 units in 100 mL NS). Polyvinyl chloride (PVC) infusion systems, including tubing, must be used.
Insulin lispro:
Must be diluted with NS to achieve a final concentration of 0.1 to 1 unit/mL (100 units of insulin [1 mL] to 100 mL of NS yields 1 unit/mL).
Storage: Vials:
Store unopened vials in refrigerator. A vial that is in use may be stored at RT for 28 days (insulin aspart, insulin glulisine, and insulin lispro) or 31 days (regular insulin). Protect from sunlight and freezing.
Infusions:
Some insulin will be initially adsorbed to the material of the infusion bag. Regular insulin and insulin lispro: Infusion bags prepared as indicated in dilution are stable for 48 hours refrigerated and then may be used at RT for up to an additional 48 hours. Insulin glulisine: Infusion bags prepared as indicated in dilution are stable at RT for 48 hours. Insulin aspart: Infusion bags prepared as indicated in dilution are stable at RT for 24 hours.
Compatibility (underline indicates conflicting compatibility information)
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
Manufacturer states, “Insulin glulisine is stable (compatible) only with NS; do not use other solutions for infusion and do not mix with other insulins for IV administration.”
One source suggests the following compatibilities:
These compatibilities refer to regular insulin only; information on specific compatibilities for insulin aspart, insulin glulisine, and insulin lispro are not available.
Additive:
Meropenem (Merrem IV).
Y-site:
Amiodarone (Nexterone), ampicillin, ampicillin/sulbactam (Unasyn), aztreonam (Azactam), caspofungin (Cancidas), cefazolin (Ancef), cefepime (Maxipime), cefotetan, ceftaroline (Teflaro), ceftazidime (Fortaz), digoxin (Lanoxin), dobutamine, doripenem (Doribax), doxapram (Dopram), esmolol (Brevibloc), famotidine (Pepcid IV), gentamicin, heparin, 6% hydroxyethyl starch (Voluven), imipenem-cilastatin (Primaxin), indomethacin (Indocin IV), levofloxacin (Levaquin), magnesium sulfate, meperidine (Demerol), meropenem (Merrem IV), midazolam (Versed), milrinone (Primacor), morphine, nitroglycerin IV, nitroprusside sodium, oxytocin (Pitocin), pantoprazole (Protonix IV), pentobarbital (Nembutal), propofol (Diprivan), sodium bicarbonate, tacrolimus (Prograf), ticarcillin/clavulanate (Timentin), tobramycin, vancomycin, vasopressin.
Rate of administration
Each 50 units or fraction thereof over 1 minute. When given in an IV infusion, the rate should be ordered by the physician and will depend on insulin and fluid needs; see Dilution for example. Decrease rate when plasma glucose reaches 300 mg/dL. Manufacturer recommends that the rate of insulin lispro be regularly adjusted to maintain blood glucose concentrations between 100 and 160 mg/dL.
Actions
A hormone produced by the pancreas that controls the storage and metabolism of carbohydrates, proteins, and fat. Responsible for regulation of glucose metabolism. Binds to insulin receptors on muscle and fat cells and lowers blood glucose by facilitating cellular uptake of glucose and inhibiting output of glucose from the liver. Also inhibits lipolysis, proteolysis, and gluconeogenesis and enhances protein synthesis and conversion of excess glucose into fat. Rapidly and widely distributed. The glucose-lowering activities of regular insulin, insulin aspart, insulin glulisine, and insulin lispro are equipotent when administered by the IV route. The average elimination half-life is dose dependent and ranges from 0.25 to 1 hour depending on dose and product.
Indications and uses
Regular insulin:
Treatment of diabetes mellitus (Type 1, Type 2, and gestational). ■ Treatment of complications associated with diabetes (e.g., diabetic coma, hyperosmolar hyperglycemia with or without coma [hyperglycemia and dehydration], ketoacidosis with or without coma [plasma glucose exceeding 250 mg/dL with arterial pH of 7 to 7.24 or less and serum bicarbonate of 10 to 15 mEq/L or less]). ■ In combination with glucose to treat hyperkalemia. ■ Add to total parenteral nutrition to control hyperglycemia.
Humulin R U-100, insulin aspart, insulin glulisine, and insulin lispro:
Treatment of patients with diabetes mellitus to improve glycemic control. Usually used as part of a SC injection regimen or as a SC infusion administered via an external insulin pump. May be given IV. Proper medical supervision is required to prevent hypoglycemia and hypokalemia.
Unlabeled uses:
Diagnosis of growth hormone deficiency. ■ Continuous intravenous infusions administered via a special pump have been used to treat severe brittle diabetics who have failed more conventional therapy.
Contraindications
Contraindicated during episodes of hypoglycemia and in patients hypersensitive to regular human insulin, insulin aspart, insulin glulisine, insulin lispro, or one of the excipients of any of these products.
Precautions
Only regular insulin, insulin aspart, insulin glulisine, and insulin lispro may be given IV. All insulin formulations for IV use are standardized at 100 units/mL.
Regular insulin, insulin aspart, insulin glulisine, and insulin lispro:
Any change in insulin should be made cautiously and only under medical supervision. Concomitant oral antidiabetic treatment may need to be adjusted. ■ Hypoglycemia and hypokalemia are potential side effects of insulin therapy. Use caution in patients in whom these side effects may be clinically relevant (e.g., patients who are fasting, have autonomic neuropathy, are using potassium-lowering drugs [e.g., diuretics], or are taking drugs sensitive to serum potassium levels [e.g., digoxin]). ■ Severe hypoglycemia may lead to unconsciousness and/or convulsions and may result in temporary or permanent impairment of brain function or death. Early warning symptoms of hypoglycemia may be different or less pronounced under certain conditions such as long-standing diabetes, diabetic nerve disease, and the use of medications such as beta-blockers. ■ Untreated hypokalemia may cause respiratory paralysis, ventricular arrhythmia, and death. ■ Use with caution in patients with renal or hepatic impairment; see Dose Adjustments. Frequent monitoring may be required. ■ Insulin requirements may be altered during illness or stress. ■ Anti-insulin antibodies have been reported. Clinical significance of anti-insulin antibodies is unknown. ■ Systemic hypersensitivity reactions have been reported. May include hypotension, pruritus, rash, shortness of breath, sweating, tachycardia, and/or wheezing. Anaphylaxis has occurred. ■ Use with thiazolidinediones (TZDs [e.g., pioglitazone (Actos)]) may cause heart failure. See Drug/Lab Interactions.
Regular insulin:
Insulin potency may be reduced by at least 20% and possibly up to 80% via the glass or plastic infusion container and plastic IV tubing before it actually reaches the venous system when given by infusion. The percentage adsorbed is inversely proportional to the concentration of insulin (the larger the dose, the less adsorption) and takes place within 30 to 60 minutes. Albumin is sometimes added to reduce this adsorption. Other additives (e.g., electrolytes, other drugs, vitamins) may also reduce adsorption. Other methods of compensation for insulin loss include the addition of added insulin to saturate binding sites or the use of a syringe pump (instead of infusion containers) to reduce surface area for adsorption.
Monitor:
Response to insulin measured by blood glucose, blood pH, acetone, BUN, sodium, potassium, chloride, and CO2 levels. Monitor patient carefully in all situations. ■ Glucose and potassium levels must be monitored closely during IV administration of insulin to avoid potentially fatal hypoglycemia and hypokalemia. ■ Glycosylated hemoglobin (HgbA1c) may be measured to assess long-term glycemic control. ■ Hypovolemia is a common complication of diabetic acidosis. ■ See Drug/Lab Interactions. ■ Insulin is inactivated at pH above 7.5.
Patient education:
Monitor blood glucose as directed. ■ Adhere to consistent diet and exercise programs. ■ Avoid alcohol. ■ Review medications or changes in medication regimen with a health care professional. ■ Review S/S of hypoglycemia and hyperglycemia with a health care professional. Be familiar with the treatment for each. ■ Insulin requirements may change with onset of illness. Monitor glucose carefully and adjust insulin therapy as required.
Maternal/child:
Regular insulin:
Category B: human insulin is drug of choice for control of diabetes in pregnancy. Additional insulin may be required to control serum glucose and avoid ketoacidosis. Monitor carefully; insulin requirements may drop immediately postpartum. Normal prepregnancy dose should be achieved within 6 weeks. Patients with gestational diabetes usually do not require insulin therapy following childbirth. ■ Use caution in breast-feeding. ■ Breast-feeding may decrease insulin requirements. ■ Inadequately controlled maternal blood glucose late in pregnancy may cause increased insulin production in the fetus. Monitor and treat neonatal hypoglycemia postpartum.
Insulin aspart and insulin lispro:
Category B: careful monitoring of glucose control is essential; see comments under regular insulin. ■ Use caution during breast-feeding.
Insulin glulisine:
Category C: use caution and assess risk versus benefit. Careful monitoring of glucose control is essential; see comments under regular insulin. ■ Use caution with breast-feeding. Considered compatible, but women with diabetes who are lactating may require adjustments of their insulin doses. ■ Safety and effectiveness of IV administration for use in pediatric patients not established.
Drug/lab interactions
Hypoglycemic effect is potentiated by ACE inhibitors (e.g., enalapril [Vasotec], lisinopril [Zestril]), anabolic steroids (e.g., nandrolone [Durabolin]), disopyramide (Norpace), fluoxetine (Prozac), fibrates (e.g., fenofibrate [Tricor]), guanethidine, MAO inhibitors (e.g., selegiline [Eldepryl]), oral antidiabetic agents (e.g., glyburide [DiaBeta]), pentoxifylline (Trental), salicylates, sulfonamides, and many others. ■ Drugs that may reduce blood glucose–lowering effect include atypical antipsychotic medications (e.g., olanzapine [Zyprexa], clozapine [Clozaril]), corticosteroids, danazol (Cyclomen), diazoxide, diuretics (e.g., furosemide [Lasix]), glucagon, isoniazid (INH), niacin, oral contraceptives, phenothiazine derivatives (e.g., chlorpromazine [Thorazine]), protease inhibitors (e.g., indinavir [Crixivan]), somatropin (Humatrope), sympathomimetic agents (e.g., dobutamine, epinephrine [Adrenalin], albuterol [Ventolin]), thyroid preparations, and others. ■ Alcohol, beta-adrenergic blockers including ophthalmics (e.g., propranolol, metoprolol [Lopressor]), clonidine (Catapres), and lithium may either potentiate or inhibit the blood glucose–lowering effect of insulin. ■ Hypoglycemic effects may be decreased in smokers; dose adjustment may be required. ■ Will affect serum potassium levels; use caution in patients taking digoxin. ■ Octreotide (Sandostatin) may alter insulin, glucagon, and growth hormone secretion, resulting in hypoglycemia or hyperglycemia. Monitor serum glucose and adjust insulin dose as indicated. Octreotide also markedly increases adsorption of insulin to glass and plastic and reduces availability. ■ Pentamidine is toxic to the beta cells of the pancreas. Patients may develop hypoglycemia initially as insulin is released. This may be followed by hypoinsulinemia and hyperglycemia with continued pentamidine therapy. ■ S/S of hypoglycemia may be masked in the presence of beta-blockers, clonidine (Catapres), guanethidine (Ismelin), and reserpine. ■ Thiazolidinediones (TZDs [e.g., pioglitazone (Actos)]) can cause dose-related fluid retention, particularly when used in combination with insulin. Fluid retention may lead to or exacerbate heart failure. Patients receiving concomitant therapy should be monitored for S/S of heart failure. Dose reduction or discontinuation of the TZD may be indicated if heart failure develops.
Side effects
Hypoglycemia with overdose. Mild to moderate: Abnormal behavior; anxiety; blurred vision; depressed mood; dizziness; drowsiness; headache; hunger; inability to concentrate; irritability; light-headedness; palpitation; personality changes; restlessness; sleep disturbances; slurred speech; sweating; tingling in the hands, feet, lips, or tongue; tremors; unsteady movement.
Severe: Coma, disorientation, hypokalemia (with ECG changes), seizures, unconsciousness, and death. Hypersensitivity reactions, including anaphylaxis, may occur; death is rare. Abdominal pain, chest pain, diarrhea, headache, hyporeflexia, nausea, and sensory disturbances have also been reported.
Antidote
Discontinue the drug immediately and notify physician of adverse reactions or hypoglycemia. Glucagon 1 to 2 mg IM or SC is the specific antidote for insulin overdose. It may be supplemented by glucose 50% IV and/or oral carbohydrates such as glucose gel or orange juice. Oral carbohydrates may be sufficient to combat early symptoms of hypoglycemia. Correct hypokalemia as indicated. Hypersensitivity reactions usually respond to symptomatic treatment.
Interferon alfa-2b, recombinant 
(in-ter-FEER-on AL-fah)
Intron A
Biologic response modifier
Antineoplastic
pH 6.9 to 7.5
Usual dose (international units [IU])
Pretreatment with acetaminophen may lessen flu-like side effects. To enhance tolerability, injections should be administered in the evening when possible. Not all dosage forms and strengths are appropriate for some indications; see Dilution.
Malignant melanoma:
Induction:
20,000,000 IU/M2 as an infusion for 5 consecutive days per week for 4 weeks. Begin therapy within 56 days of surgical resection.
Maintenance:
Follow with 10,000,000 IU/M2 as a SC injection three times each week for 48 weeks.
Total treatment regimen lasts for 1 year and should be completed unless there is progression of disease or the drug is discontinued because of specific side effects.
Dose adjustments
Moderate to severe adverse events may require modification of the dosage regimen or, in some cases, termination of therapy. Temporarily discontinued for serious adverse reactions, if granulocytes decrease to less than 500/mm3, or ALT/AST increases to 5 to 10 times the ULN. When adverse reactions subside or improvement of granulocytes or liver function tests occurs, treatment can be restarted at 50% of the previous dose. Discontinue permanently if serious adverse reactions persist, if a severe adverse reaction recurs at a reduced dose, if granulocytes decrease to less than 250/mm3, if platelet counts decrease to less than 25,000/mm3, or if ALT/AST increases to more than 10 times the upper limit of normal. ■ See Elderly.
Dilution (international units [IU])
Intron A Solution for Injection is NOT recommended for IV administration and should not be used for the induction phase of malignant melanoma. Only the sterile powder is suitable for dilution for IV use. Do not use different brands of interferon in any single treatment regimen; variations exist and may adversely affect dosage and response to treatment. Powder for Injection is available in 10, 18, and 50 million IU vials; select one most appropriate for desired dose. Allow product to come to RT before use. Reconstitute immediately before use with 1 mL of SWFI (provided). (The SWFI supplied by the manufacturer contains 5 mL. Discard remaining 4 mL.) Gently swirl to dissolve. Withdraw desired dose and inject into 100 mL of NS. Final concentration should be at least 10 million IU/100 mL.
Storage:
Unopened vials of powder for injection should be refrigerated. After reconstitution, solution should be used immediately but can be refrigerated at 2° to 8° C (36° to 46° F) for 24 hours. Single-dose vial. Discard any unused portion.
Compatibility
Specific information not available. Consider specific use; consult pharmacist. One source indicates compatibility with NS, LR, and R solutions and incompatibility with dextrose solutions.
Rate of administration
A single dose equally distributed over 20 minutes. Administration at bedtime and the use of acetaminophen may prevent or partially reduce common side effects (e.g., fever, headache, “flu-like” symptoms).
Actions
A naturally occurring small protein and glycoprotein of the interferon family produced by recombinant DNA techniques. It binds to specific membrane receptors on the cell surface and initiates a complex sequence of intracellular events (e.g., induction of specific enzymes, suppression of cell proliferation, immunomodulating activities [e.g., enhancement of phagocytic activity of macrophages, augmentation of the specific cytotoxicity of lymphocytes for target cells, inhibition of virus replication in virus-infected cells]). Peak concentration achieved within 30 minutes. Half-life about 2 hours; undetectable 4 hours after infusion. The kidney may be the site of interferon catabolism. Has produced a significant increase in relapse-free and overall survival in patients with malignant melanoma.
Indications and uses
Adjuvant to surgical treatment in patients with malignant melanoma who are free of disease but at high risk for systemic recurrence within 56 days of surgery. ■ Used IM or SC to treat hairy cell leukemia, follicular non-Hodgkin’s lymphoma, AIDS-related Kaposi’s sarcoma, chronic hepatitis B, chronic hepatitis C, and intralesionally to treat condylomata acuminata.
Contraindications
Hypersensitivity to interferon alfa or any of its components. ■ Decompensated liver disease or autoimmune hepatitis.
Precautions
May cause or aggravate life-threatening or fatal neuropsychiatric (e.g., depression, aggressive and/or suicidal behavior), autoimmune, ischemic, or infectious disorders. Close monitoring with periodic clinical and laboratory evaluations required. Discontinue therapy if S/S of any of the previous conditions are persistently severe or worsen. Symptoms resolve in most cases. ■ Because of fever and other flu-like symptoms associated with its use, interferon alfa should be used cautiously in patients with a history of pulmonary disease (e.g., COPD), diabetes mellitus prone to ketoacidosis, coagulation disorders (e.g., thrombophlebitis, pulmonary embolism), or severe myelosuppression. ■ Use cautiously in patients with a history of cardiovascular disease (e.g., unstable angina, recent MI, and previous or current arrhythmic disorder). Cardiovascular adverse events (e.g., arrhythmia, cardiomyopathy, hypotension, MI, tachycardia) have been reported. Supraventricular arrhythmias occurred rarely and appeared to be correlated with pre-existing conditions or prior therapy with cardiotoxic agents. ■ Depression and suicidal behavior (including suicidal ideation, suicidal attempts, and completed suicides), homicidal ideation, and aggressive behavior sometimes directed toward others have been reported; see Monitor and Maternal/Child. ■ Use with caution in patients with a history of psychiatric disorders and in patients with substance use disorders. Drug screening and periodic health evaluation, including psychiatric symptom monitoring, may be indicated. ■ Severe cytopenias, including rare cases of aplastic anemia, have been reported; see Monitor. ■ Ophthalmologic disorders (e.g., decrease or loss of vision; retinopathy, including macular edema, retinal artery or vein thrombosis, retinal hemorrhage, and cotton wool spots; optic neuritis; papilledema; and serous retinal detachment) may be induced or aggravated by treatment with interferons; see Monitor. ■ Use caution in patients with psoriasis and sarcoidosis; may exacerbate disease. ■ Rare cases of autoimmune diseases (e.g., lupus erythematosus, Raynaud’s phenomenon, rhabdomyolysis, rheumatoid arthritis, thrombocytopenia, and vasculitis) have been reported; may require discontinuation of therapy. ■ Serum neutralizing antibodies have been detected in some patients; clinical significance unknown. ■ Ischemic and hemorrhagic cerebrovascular events have been reported. A causal relationship between interferon therapy and these side effects is difficult to establish. ■ Endocrine disorders (e.g., hypothyroidism, hyperthyroidism, diabetes mellitus) have developed in patients receiving interferon alfa-2b. ■ Hepatotoxicity, including fatality, has been reported. Interferon alfa increases the risk of hepatic decompensation and death in patients with cirrhosis. ■ Pulmonary disorders (e.g., dyspnea, pulmonary infiltrates, pneumonia, bronchiolitis obliterans, interstitial pneumonitis, pulmonary hypertension, and sarcoidosis), some resulting in respiratory failure and/or patient deaths, may be induced or aggravated by interferons. Recurrence of respiratory failure has been observed with interferon rechallenge. ■ Acute serious hypersensitivity reactions have been reported. ■ Elevated triglyceride levels have been observed. Hypertriglyceridemia may result in pancreatitis.
Monitor:
Obtain baseline CBC including differential and platelet count, blood chemistries (electrolytes, glucose, liver function tests, and TSH), and chest x-ray. Obtain baseline eye exam for all patients and periodically for patients with pre-existing ophthalmologic disorders (e.g., diabetic or hypertensive retinopathy). Obtain baseline ECG if there is a history of cardiovascular disease and/or patient is in advanced stages of cancer. Monitor all tests periodically during therapy. Adjust or discontinue therapy as appropriate. ■ In patients with malignant melanoma, monitor CBC with differential and platelet count and liver function tests weekly during the induction phase and monthly during maintenance. Dose adjustments will be determined by results. Discontinue therapy if neutrophil count drops below 500 cells/mm3. ■ Hepatic function should be monitored in all patients receiving interferon. Obtain serum bilirubin, ALT, AST, alkaline phosphatase, and LDH at 2, 8, and 12 weeks following initiation of therapy and then every 6 months while on therapy. ■ Monitor for thrombocytopenia (platelet count less than 50,000/mm3). Discontinue therapy if platelet count drops below 25,000/mm3. Initiate precautions to prevent excessive bleeding (e.g., inspect IV sites, skin, and mucous membranes; use extreme care during invasive procedures; test urine, emesis, stool, and secretions for occult blood). ■ Monitor for S/S of a hypersensitivity reaction (e.g., anaphylaxis, angioedema, bronchoconstriction, urticaria). ■ Closely monitor any patient with a history of cardiovascular disease (see Precautions); arrhythmias (including supraventricular arrhythmias and tachycardia), hypotension, and transient reversible cardiomyopathy have occurred. ■ Hypotension may occur during administration or up to 2 days after therapy; monitor BP frequently. May require fluid replacement to maintain intravascular volume. ■ Maintain adequate hydration, particularly during initial states of treatment. ■ Monitor for signs of depression and/or aggressive or suicidal behavior. If psychiatric problems develop, patients should be carefully monitored during treatment and in the 6-month follow-up period. If psychiatric symptoms persist or worsen or if suicidal or homicidal ideation or aggressive behavior toward others is identified, treatment with interferon alpha should be discontinued. The patient should be followed closely, with psychiatric intervention as appropriate. Use of narcotics, hypnotics, or sedatives may be required to manage adverse effects; use with caution and monitor carefully. ■ Monitor thyroid function, liver function tests, blood glucose, and triglyceride levels; abnormalities not normalized by medication may require discontinuing interferon. ■ Determine cause of persistent fever (not thought to be due to the flu-like syndrome commonly reported in patients treated with interferon alfa). ■ Repeat chest x-ray if cough, dyspnea, fever, or other respiratory symptoms occur. Monitor closely if pulmonary infiltrates or evidence of pulmonary function impairment is present; may require discontinuing interferon. ■ Obtain an eye exam for any patient who complains of changes in visual acuity, visual fields, or other ophthalmologic symptoms. ■ See Precautions.
Patient education:
Cooperation for close monitoring and prompt reporting of side effects is imperative. Side effects may include depression (including suicidal ideation), cardiovascular toxicity (e.g., chest pain), ophthalmologic toxicity (e.g., loss of or change in vision), pancreatitis (abdominal pain), cytopenias (persistently high fevers, bruising). ■ Review manufacturer’s medication guide before each dose. ■ Home use may require extensive patient education. Review Instructions for Use provided by manufacturer. ■ Do not change brands of interferon without medical consultation. Variations in dosage, routes of administration, and adverse reactions exist among different brands. ■ Do not drive or operate machinery; mental alertness may be impaired. ■ Administration at bedtime and/or use of acetaminophen may be helpful to prevent the most common flu-like side effects. Remain well hydrated. ■ Side effects may decrease in severity as treatment continues. ■ See Appendix D, p. 1333.
Maternal/child:
Category C: has been shown to have abortifacient effects in monkeys. Should be used only if benefits justify risks. ■ Discontinue breast-feeding. ■ Safety for use in pediatric patients under 18 years of age for indications other than chronic hepatitis B or C has not been established. ■ Suicidal ideation or attempts have occurred more often among pediatric patients (primarily adolescents) compared to adult patients.
Elderly:
Incidence of encephalopathy, obtundation, and coma has occurred, especially at higher doses. ■ Cardiovascular adverse events and confusion have been reported more frequently in the elderly. ■ Consider age-related decreases in organ function and concomitant disease and drug therapy. Careful monitoring required. Make dose adjustments based on symptoms and/or lab abnormalities.
Drug/lab interactions
Specific information not available. Use caution with any other potentially myelosuppressive agent (e.g., cisplatin, zidovudine [AZT, Retrovir]). ■ May increase serum levels of theophylline; monitoring of theophylline levels recommended. ■ See Precautions.
Side effects
Occur in high percentages of patients; may be serious enough to require a decreased dose or discontinuation of drug. Usually rapidly reversible after therapy is discontinued, but may require up to 3 weeks to resolve. The most frequently reported adverse reactions were flu-like symptoms (chills, fatigue, fever, headache, and myalgia). In patients treated for malignant melanoma, the most frequently reported adverse reaction was fatigue. Adverse reactions that were reported in greater than 20% of patients included alopecia, altered taste sensation, anemia, anorexia, chills, depression, diarrhea, dizziness/vertigo, fever, headache, increased AST, myalgia, nausea/vomiting, and neutropenia. Severe adverse reactions (ECOG [Eastern Cooperative Oncology Group] Toxicity Criteria Grade 3 or 4) reported in greater than 10% of patients included chills, fatigue, fever, headache, increased AST, myalgia, and neutropenia/leukopenia. Many other side effects may occur.
Post-marketing:
Stevens-Johnson syndrome, toxic epidermal necrolysis, erythema multiforme, injection site necrosis, myositis, hearing loss, pulmonary fibrosis, and a wide variety of autoimmune and immune-mediated disorders (e.g., idiopathic thrombocytopenic purpura and thrombotic thrombocytopenic purpura) have been identified in post-marketing reports.
Antidote
Keep physician informed of all side effects. Some will be tolerated or treated symptomatically. Discontinue interferon immediately for any acute serious hypersensitivity reactions and treat as appropriate; may require epinephrine, airway management, oxygen, IV fluids, antihistamines (e.g., diphenhydramine [Benadryl]), corticosteroids (e.g., hydrocortisone sodium succinate [Solu-Cortef]), and pressor amines (e.g., dopamine). Discontinue drug for any patient who develops severe depression or other psychiatric disorder. Close patient monitoring and psychiatric intervention may be indicated. Discontinue for myelosuppression that persists after dose reduction, endocrine abnormalities or persistently elevated triglycerides associated with symptoms of potential pancreatitis (e.g., abdominal pain, nausea, vomiting) not normalized by medication, serious liver function abnormalities (Grade 3 hepatic injury or hepatic decompensation [Child-Pugh score >6 (Class B and C)]), new or worsening ophthalmologic disorders, serious pulmonary function impairment, or pulmonary infiltrates. Treatment of overdose with hemodialysis or peritoneal dialysis is not considered effective. Resuscitate as necessary.
Ipilimumab 
(ip-i-LIM-ue-mab)
Yervoy
Monoclonal antibody
Antineoplastic
pH 7
Usual dose
Unresectable or metastatic melanoma:
3 mg/kg administered as an IV infusion over 90 minutes every 3 weeks for a total of 4 doses. In the event of toxicity, doses may be delayed, but all treatment must be administered within 16 weeks of the first dose.
Adjuvant treatment of melanoma:
10 mg/kg as an IV infusion over 90 minutes every 3 weeks for 4 doses followed by 10 mg/kg every 12 weeks for up to 3 years. In the event of toxicity, doses are omitted, not delayed.
Dose adjustments
The recommended treatment modifications for immune-mediated adverse reactions to ipilimumab are outlined in the following chart.
| Treatment Modifications of Ipilimumab for Immune-Mediated Adverse Reactions | ||
| Target/Organ System | Adverse Reaction (CTCAE, v3) | Treatment Modification |
| Endocrine | Symptomatic endocrinopathy | Withhold ipilimumab. Resume ipilimumab in patients with complete or partial resolution of adverse reactions (Grade 0 or 1) and who are receiving less than 7.5 mg prednisone or equivalent per day. |
• Symptomatic reactions lasting 6 weeks or longer • Inability to reduce corticosteroid dose to 7.5 mg prednisone or equivalent per day Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

| ||