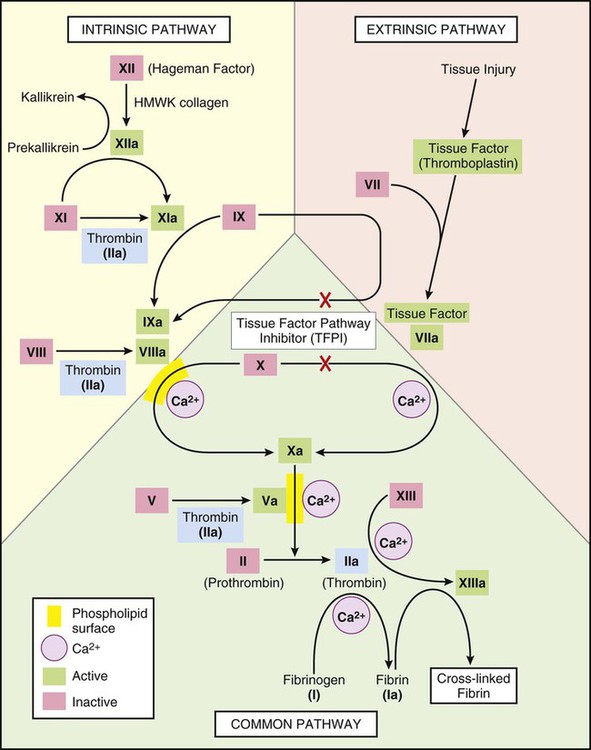
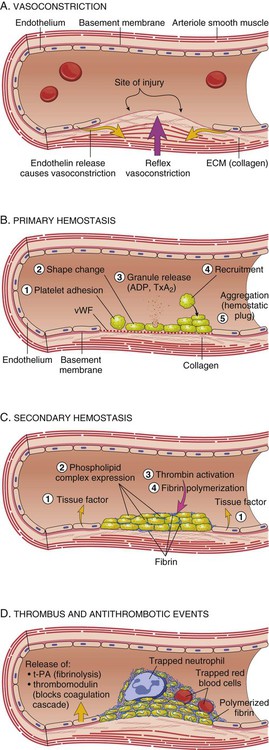
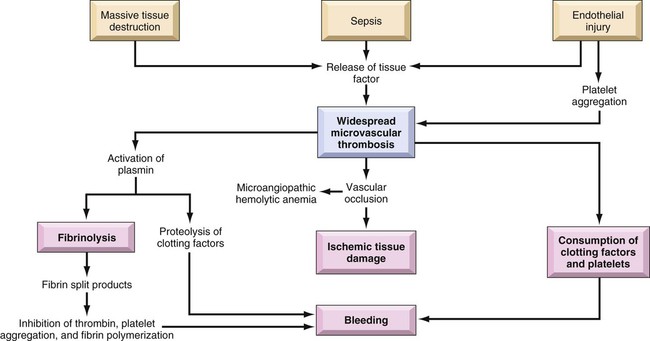
Hemostasis, the ability of the body to control bleeding and clotting, is an intricate balancing act between the coagulation mechanism and fibrinolysis. Four major actions are involved in achieving hemostasis: 1) local vasoconstriction to reduce blood flow; 2) platelet aggregation at the injury site and formation of a platelet plug; 3) formation of a fibrin mesh to strengthen the plug; and 4) dissolution of the clot after tissue repair is complete.1 Disruption of the normal hemostatic balance can result in devastating hemorrhagic or thrombotic conditions. The coagulation mechanism consists of 13 factors that work together through a series of feedback loops to achieve hemostasis (Table 38-1 and Fig. 38-1). Depending on the initial triggering event, the extrinsic or intrinsic coagulation pathway is intiated.1,2 The extrinsic pathway begins when vascular injury occurs, resulting in release of tissue factor and activation of coagulation factor VII. The intrinsic pathway is activated when the damaged subendothelium comes into direct contact with circulating blood. In this contact phase, proteins activate additional coagulation factors (XII, XI, IX, and VIII).1 At this point, the two pathways converge into a common pathway, and prothrombin and fibrinogen are converted to their active forms, resulting in clot formation.3,4 TABLE 38-1 COAGULATION FACTORS AND COMMON NAMES Platelets are activated by the arrival of thrombin at the site of injury (Fig. 38-2). Local platelets change shape, become sticky, and begin to aggregate along the vessel wall. Activated platelets undergo degranulation, releasing several factors to assist in clot formation. Serotonin and histamine, two potent vasoconstrictors, help limit blood loss while the clot is forming. The prostaglandin thromboxane A2 (TXA2) contributes to vasoconstriction and promotes further platelet degranulation. Adenosine diphosphate (ADP) recruits platelets by increasing adherence and degranulation,2,3 and the process continues. At the convergence of the intrinsic and extrinsic pathways, factor X is converted into its active form, thereby enabling the conversion of prothrombin to thrombin. Thrombin then converts fibrinogen to fibrin. Strands of fibrin form and radiate around the newly formed clot, essentially creating a net in which platelets, red blood cells (RBCs), and white blood cells (WBCs) are trapped.1 The clot is further secured to the site as platelet actomyosin causes it to contract and consolidate.2 Under normal conditions, feedback systems prevent the coagulation process from spinning out of control. Prostacyclin I2 (PGI2) is a prostaglandin released from damaged endothelial cells. It counteracts the effects of TXA2, serotonin, and histamine through vasodilation and inhibition of platelet degranulation.1–3 Another means of regulating clot formation is through inhibition of enzymes necessary for activation of coagulation factors along the intrinsic and extrinsic pathways, preventing the conversion of prothrombin to thrombin. The most important of these regulators is antithrombin III; however, protein C and protein S also play a role in thrombin inhibition.5 The process of fibrinolysis promotes dissolution and remolding of the clot to promote repair of the vessel wall and maintain flow through the vessel lumen (Fig. 38-2D). Fibrinolysis begins as soon as the fibrin clot is formed. Circulating plasminogen, a precursor to the powerful enzyme plasmin, binds to fibrin and is trapped within the newly formed clot. The injured epithelial wall releases tissue-type plasminogen activator (tPA), which converts plasminogen to its active state, plasmin. The plasmin begins to digest the fibrin, rapidly breaking down the clot.2,4 When fibrin is broken down, the fibrin degradation products released act as anticoagulants. Disseminated intravascular coagulation (DIC) is a syndrome that arises as a complication of other serious or life-threatening conditions. Although DIC is not seen often, it can seriously hamper diagnostic and treatment efforts for the critically ill patient. An understanding of the etiologic and pathophysiologic mechanisms of DIC can assist in anticipating the syndrome’s occurrence, recognizing its signs and symptoms, and prompting intervention. Also known as consumptive coagulopathy, DIC is characterized by bleeding and thrombosis, both of which result from depletion of clotting factors, platelets, and RBCs. If not treated quickly, DIC will progress to multiple organ failure and death.6 Many clinical events can prompt the development of DIC in the critically ill patient, but the exact underlying trigger may not be identifiable (Box 38-1). There are, however, some commonly known conditions associated with the development of DIC. Sepsis, particularly that caused by gram-negative organisms, can be identified as the culprit in as many as 20% of cases, making it the most common cause of DIC. In this instance, endotoxins serve as a trigger for activation of tissue factor and the extrinsic coagulation pathway. Metabolic acidosis and hypoperfusion associated with shock syndromes can result in increased formation of free radicals and damage to tissues. Tissue factor is activated, resulting in DIC. Massive trauma or burns are frequently associated with DIC. Direct tissue damage activates the extrinsic coagulation pathway, and damage to endothelial surfaces activates the intrinsic pathway.3 Obstetric emergencies, such as abruptio placenta, retained placenta, or incomplete abortion, are also associated with the development of DIC. Tissue factor is concentrated in the placenta, and damage or disruption of this structure can activate coagulation pathways, resulting in coagulopathy.7 Regardless of the cause, the common thread in the development of DIC is damage to the endothelium that results in activation of the coagulation mechanism (Fig. 38-3).1 The extrinsic coagulation pathway plays a major role in the development of DIC. Direct damage to the endothelium results in the release of tissue factor and activation of this pathway. The secondary surge of thrombin formation as a result of activation of the intrinsic coagulation pathway leads to the massive disruption of the delicate balance that is hemostasis. Excessive thrombin formation results in rapid consumption of coagulation factors and depletion of regulatory substances—protein C, protein S, and antithrombin.5 With no checks and balances, thrombi continue to form along damaged epithelial walls, resulting in occlusion of the vessels. As occlusion reaches a critical level, tissue ischemia ensues, leading to further tissue damage and perpetuating the process. Eventually, end-organ function is affected by the ischemia, and failure is evident.1 In response to the formation of clots, the fibrinolytic system is activated. As plasmin breaks down the fibrin clots, fibrin split products are released, and they act as anticoagulants.3,7 Coupled with depletion of circulating clotting factors, activation of fibrinolysis results in excessive bleeding. The end result is shock and further tissue ischemia that aggravate end-organ dysfunction and failure. Death is imminent if this destructive cycle is not interrupted.8 Clinical manifestations are related to the two primary pathophysiologic mechanisms of DIC: the formation of thrombi and bleeding. Thrombi in peripheral capillaries can lead to cyanosis, particularly in the fingers, toes, ears, and nose. In severe, untreated cases, this peripheral ischemia may progress to gangrene.3,6 As the condition progresses, ischemia worsens, and end organs are affected. The result of this more central ischemia can be respiratory insufficiency and failure, acute kidney injury, bowel infarction, and ischemic stroke. The tissue damage that results perpetuates the anomalies of DIC.3 As coagulation factors are depleted, bleeding from intravenous and other puncture sites is observed. Ecchymoses may result from even routine interventions such as the use of a manual blood pressure cuff, bathing, or turning.3,6 Bloody drainage may also occur from surgical sites, drains, and urinary catheters. With progression of DIC, the patient is at risk for severe gastrointestinal or subarachnoid hemorrhage.3,6 Table 38-2 lists many of the common signs and symptoms of DIC. TABLE 38-2 COMMON SIGNS AND SYMPTOMS OF DISSEMINATED INTRAVASCULAR COAGULATION Laboratory tests used to diagnose DIC essentially assess the four basic characteristics of this syndrome: 1) increased coagulant activity; 2) increased fibrinolytic activity; 3) impaired regulatory function; and 4) end-organ failure.3 Continuous activation of the coagulation pathways results in consumption of coagulation factors. Because of this, the prothrombin time (PT), the activated partial thromboplastin time (aPTT), and the international normalized ratio (INR) values are elevated. Although the platelet count may fall within normal ranges, serial examination reveals a declining trend in values. An unexpected drop of at least 50% in the platelet count, particularly in the presence of known contributing factors and associated signs and symptoms, strongly indicates DIC.1 Fibrinogen levels drop as more and more clots are formed. Thrombus formation in small vessels narrows the vessel lumen, forcing RBCs to squeeze through. The resulting damage and fragmentation of these cells can be seen on microscopic examination of blood samples. Damaged, fragmented RBCs are called schistocytes.3,6 In response to the excess clotting activity, the fibrinolytic process accelerates, and levels of byproducts increase. This is reflected in markedly elevated levels of fibrin degradation products. Another key laboratory test used to evaluate the degree of clot dissolution—and therefore the severity of the coagulopathy—is the d-dimer level.1 d-dimers exclusively indicate clot degradation because, unlike fibrin degradation products, which also result from the breakdown of free circulating fibrin, d-dimers result only from dissolution of clots.3 With progression of the coagulopathy, normal regulatory mechanisms are disrupted, as reflected in decreasing levels of inhibitory factors such as protein C, factor V, and antithrombin III.3,6,9 Unchecked DIC resulting in occlusion of vessels and tissue ischemia leads to end-organ dysfunction. Respiratory failure, indicated by abnormal arterial blood gas (ABG) levels; liver failure, indicated by increasing liver enzymes; and renal impairment, indicated by rising blood urea nitrogen (BUN) and creatinine levels are common findings in advanced DIC.9 No single laboratory study can confirm the diagnosis of DIC, but several key results are strong indicators of the condition (Table 38-3). The International Society of Thrombosis and Hemostasis emphasizes early detection of DIC through observation of abnormal trends in laboratory values.5 TABLE 38-3 KEY LABORATORY STUDIES IN DISSEMINATED INTRAVASCULAR COAGULATION Without question, the primary intervention in DIC is prevention. Being aware of the conditions that commonly contribute to the development of DIC and treating them vigorously and without delay provide the best defense against this devastating condition.3,6,7,9 After DIC is identified, maintaining organ perfusion and slowing consumption of coagulation factors are paramount to achieving a favorable outcome.1,3 Multiple organ dysfunction syndrome (MODS) frequently results from DIC and exacerbates the underlying pathology.8 It is essential to prevent end-organ ischemia and damage by supporting blood pressure and circulating volume. Administration of intravenous fluids and inotropic agents and, if overt hemorrhaging is evident, infusion of packed RBCs are appropriate interventions to replace blood volume and essential, oxygen-carrying RBCs. In the presence of severe platelet depletion (less than 50,000/mm3) and severe hemorrhage, platelet transfusions are often indicated.5,6 However, caution must be used when administering platelets because antiplatelet antibodies may be formed. These antibodies may become activated during future platelet transfusions and elicit DIC.3 Replacement of clotting factors in the patient with DIC is thought by some authorities to perpetuate the coagulopathy; however, there is little scientific evidence to support this theory.1 Fibrinogen levels less than 100 mg/dL indicate the appropriateness of administering cryoprecipitate. A prolonged PT indicates the need for fresh-frozen plasma.3,5,6 Slowing consumption of coagulation factors by inhibiting the processes involved in clot formation is another strategy used in treating DIC. The use of heparin, particularly low–molecular- weight heparin (LMWH), to prevent formation of future clots is controversial.6 It is contraindicated in patients with DIC associated with recent surgery or with gastrointestinal or central nervous system (CNS) bleeding. However, heparin has been beneficial in obstetric emergencies such as retained placenta or incomplete abortion, severe arterial occlusions, or MODS caused by microemboli.3,6 Inhibitors such as aminocaproic acid may be used in conjunction with heparin.3,6 The use of recombinant activated protein C is gaining popularity in treating DIC, especially in the setting of severe sepsis. Activated protein C acts as an anticoagulant and works to restore normal inhibition of coagulation pathways. However, it has been associated with an increased incidence of intracerebral bleeding and must be used with caution in patients with severely decreased platelets.3,5 Thrombin production in DIC surpasses that of antithrombins and other regulatory factors that would normally be present to inactivate thrombin and its subsequent actions. The use of antithrombin III has recently been approved in the United States. Ongoing research is yielding mixed results in the treatment of DIC.5 One interesting area of research is the use of protease inhibitors. Protease molecules normally inhibit the conversion of fibrinogen to fibrin in the coagulation mechanism, but in DIC, this inhibitory mechanism is impaired. The introduction of protease inhibitors by intravenous infusion may be advantageous in arresting DIC.3 Nursing management of the patient with DIC incorporates a variety of nursing diagnoses (Box 38-2). Assessment and monitoring are the primary weapons in the critical care nurse’s arsenal against DIC. Knowing the diseases and conditions that are most often associated with DIC and understanding the pathophysiologic mechanisms involved enables the critical care nurse to anticipate its development and intervene quickly. Nursing interventions include supporting the patient’s vital functions, initiating bleeding precautions, providing comfort and emotional support, and maintaining surveillance for complications. Disseminated Intravascular Coagulation Frequent assessments should include parameters for neurologic status, renal function, cardiopulmonary function, and skin integrity that indicate impaired tissue or organ perfusion. Particular parameters to include are mental status, BUN and creatine levels, urine output, vital signs, hemodynamic values, cardiac rhythm, arterial blood gas and pulse oximetry values, skin breakdown, ecchymoses, or hematomas.6 The critical care nurse must recognize and support the patient’s vital physiologic functions. Administration of intravenous fluids, blood products, and inotropic agents to provide adequate hemodynamic support and tissue oxygenation is essential in preventing or combating end-organ damage. Close monitoring of vital signs, hemodynamic parameters, intake and output, and appropriate laboratory values assists the critical care nurse in administering and titrating appropriate agents.3 Awareness of the patient’s bleeding potential necessitates adjustments to normal nursing interventions (Box 38-3). The nurse avoids unnecessary venipunctures that may result in bleeding, bruising, or hematomas by drawing blood from and administering medications through existing arterial or venous lines. The use of manual or automatic blood pressure cuffs is avoided whenever possible. If tracheal or oral suctioning is necessary, the use of low-level suction is recommended.6 Meticulous skin care is advised, keeping the skin moist and using specialty mattresses and beds as appropriate to prevent breakdown. Gentle care should be used when bathing or turning the patient to prevent bruising or hematoma formation. NIC Bleeding Precautions Reduction of stimuli that may induce bleeding or hemorrhage in at-risk patients Monitor the patient closely for hemorrhage Note hemoglobin/hematocrit levels before and after blood loss, as indicated Monitor for signs and symptoms of persistent bleeding (e.g., check all secretions for frank or occult blood) Monitor coagulation studies, including prothrombin time (PT), partial thromboplastin time (PTT), fibrinogen, fibrin degradation/split products, and platelet counts, as appropriate Monitor orthostatic vital signs, including blood pressure Maintain bed rest during active bleeding Administer blood products (e.g., platelets, fresh-frozen plasma), as appropriate Protect the patient from trauma, which may cause bleeding Avoid injections (IV, IM, or SQ), as appropriate Instruct the ambulating patient to wear shoes Use soft toothbrush or toothettes for oral care Use electric razor, instead of straight-edge, for shaving Tell patient to avoid invasive procedures; if they are necessary, monitor closely for bleeding Coordinate timing of invasive procedures with platelet or fresh-frozen plasma transfusions, if appropriate Refrain from inserting objects into a bleeding orifice Avoid taking rectal temperatures Administer mediations (e.g., antacids), as appropriate Instruct patient to avoid aspirin and other anticoagulants Instruct patient to increase intake of foods rich in vitamin K Use therapeutic mattress to minimize skin trauma Avoid constipation (e.g., encourage fluid intake and stool softeners), as appropriate Instruct the patient and/or family on signs of bleeding and appropriate actions (e.g., notify the nurse), should bleeding occur From Bulechek GM, et al. Nursing Interventions Classification (NIC). 6th ed. St. Louis: Mosby; 2013. Thrombocytopenia is defined as a platelet count less than 140,000/mm3.10,11 Similar to DIC, thrombocytopenia often results from another underlying condition that affects the platelet count. Thrombocytopenia is more common in women than in men, affecting adults most often between the ages of 20 and 50 years.12,13 Onset of thrombocytopenia often follows a viral infection, pregnancy, administration of certain medications (e.g., heparin, thiazide diuretics, chemotherapeutic agents), malignancies, splenomegaly, blood transfusions, or alcoholism.12 Regardless of the precipitating condition, the development of thrombocytopenia occurs by means of one of four mechanisms: 1) decreased platelet production; 2) increased platelet destruction; 3) splenic sequestration of platelets; and 4) platelet dilution.10 The most common form of thrombocytopenia seen in the critical care unit is immune thrombocytopenic purpura (ITP).11 In ITP, lymphocytes produce antibodies that begin to destroy existing platelets. The cause of this autoimmune response is unknown.13 With insufficient platelets available, the normal coagulation pathways are disrupted. Inadequate hemostasis ensues, and bleeding results. Although life-threatening gastrointestinal or intracerebral bleeding can occur, the bleeding seen in ITP does not result in deep visceral hemorrhage and hematoma.10 ITP is characterized by the gradual onset of signs and symptoms.13 The diagnosis is primarily based on findings in the patient’s history and physical examination. Petechial hemorrhages, which manifest as small red spots primarily on legs and oral mucosa, are most indicative of a platelet disorder, unlike larger hematomas, which are most commonly associated with coagulation disorders.11 Bruising unrelated to trauma is another common sign of ITP.12 The signs and symptoms of ITP are listed in Table 38-4. TABLE 38-4 IDIOPATHIC THROMBOCYTOPENIA PURPURA: SIGNS, SYMPTOMS, AND LABORATORY DATA
Hematologic Disorders and Oncologic Emergencies
Overview of Coagulation and Fibrinolysis
Coagulation Mechanism
FACTOR
COMMON NAME
I
Fibrinogen
II
Prothrombin
III
Tissue factor
IV
Calcium
V
Proaccelerin
VI
Accelerin
VII
Proconvertin
VIII
Antihemophilic
IX
Christmas factor
X
Stuart
XI
Plasma thromboplastin antecedent
XII
Hageman factor
XIII
Fibrin stabilizing factor
Clot Formation
Regulatory Mechanisms
Fibrinolysis
Disseminated Intravascular Coagulation
Description
Etiology
Pathophysiology
Assessment and Diagnosis
Clinical Manifestations
SYSTEM
SIGNS RELATED TO HEMORRHAGE
SIGNS RELATED TO THROMBI
Integumentary
Bleeding from gums, venipunctures, and old surgical sites; epistaxis; ecchymoses
Peripheral cyanosis, gangrene
Cardiopulmonary
Hemoptysis
Dysrhythmias, chest pain, acute myocardial infarction, pulmonary embolus, respiratory failure
Renal
Hematuria
Oliguria, acute kidney injury, renal failure
Gastrointestinal
Abdominal distention, hemorrhage
Diarrhea, constipation, bowel infarct
Neurologic
Subarachnoid hemorrhage
Altered level of consciousness, ischemic stroke
Laboratory Findings
TEST
VALUE
Prothrombin time (PT)
>12.5 sec
Platelets
<50,000/mm3, or at least 50% drop from baseline
Activated partial thromboplastin time (aPTT)
>40 sec
d-dimer
>250 ng/mL
Fibrin degradation products (FDP)
>40 mg/mL
Fibrinogen
<100 mg/dL
Medical Management
Nursing Management
 Box 38-2 Nursing Diagnoses
Box 38-2 Nursing Diagnoses
Supporting Patient’s Vital Functions
Initiating Bleeding Precautions
 Box 38-3
Box 38-3
Thrombocytopenia
Description
Etiology
Pathophysiology
Assessment and Diagnosis
Clinical Manifestations
SYSTEM OR STUDY
SIGNS AND SYMPTOMS
Integumentary
Petechial hemorrhage of lower extremities, ecchymoses, gingival bleeding, spontaneous epistaxis
Neurologic
Sudden, severe headache; nausea and vomiting; seizures; focal neurologic deficits; decreased level of consciousness
Renal
Hematuria
Gastrointestinal
Hematemesis, melena, hematochezia
Other
Heavy menses in women, retinal hemorrhage
Laboratory
Decreased platelet count, often <30,000 mm3 ![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Hematologic Disorders and Oncologic Emergencies
Get Clinical Tree app for offline access
