CHAPTER 8 Hematologic Disorders
Section One Disorders of the Red Blood Cells
Anemias can be classified in two ways: (1) those involving diminished production or accelerated loss of RBCs (etiology) or (2) those involving cell size (morphology). For details, see TABLES 8-1 and 8-2. Three common types of anemias are discussed in this section: anemia of chronic disease, hemolytic and sickle cell anemia, and hypoplastic (aplastic) anemia.
TABLE 8-1 ETIOLOGIC (PATHOPHYSIOLOGIC) CLASSIFICATIONS OF ANEMIA
| TYPE | CAUSE | DISEASES |
|---|---|---|
| Decreased or defective production of erythrocytes | Altered hemoglobin synthesis | Iron deficiency |
| Thalassemia | ||
| Anemia of chronic inflammation or disease | ||
| Altered DNA synthesis from deficient nutrients | Pernicious anemia (decreased vitamin B12, folate) | |
| Stem cell dysfunction | Aplastic anemia, myeloproliferative leukemia or dysplasia | |
| Bone marrow infiltration | Carcinoma, lymphoma, multiple myeloma | |
| Autoimmune disease or idiopathic | Pure red cell aplasia | |
| Increased erythrocyte destruction | Blood loss | Acute (hemorrhage, trauma) |
| Chronic (gastrointestinal bleeding, menorrhagia) | ||
| Hemolysis (intrinsic) | Hereditary spherocytosis, sickle cell trait or disease, pyruvate kinase deficiency, glucose-6-phosphate dehydrogenase (G6PD) deficiency | |
| Hemolysis (extrinsic) | Warm or cold antibody disease, infection (malarial, clostridial), erythrocyte trauma (hemolytic uremic syndrome, TTP, mechanical cardiac valve, paravalvular leak), splenic sequestration, burns |
TTP, Thrombotic thrombocytopenic purpura.
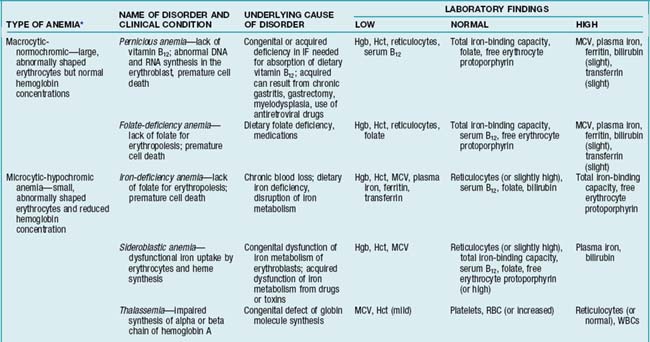
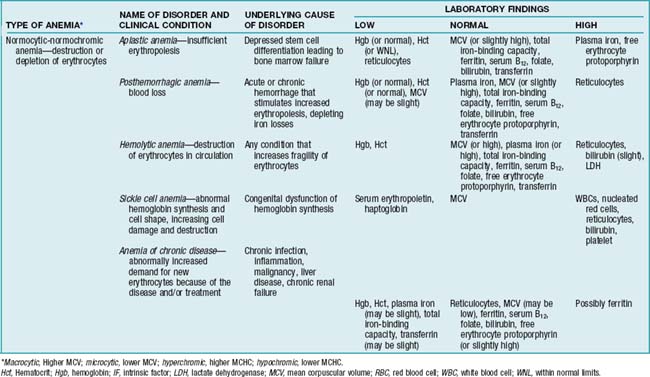
 Anemia of Chronic Disease
Anemia of Chronic Disease
Diagnostic Tests
Ferritin
Normal or increased. However, if it is less than 30 mcg/L, there is a coexisting iron deficiency.
Peripheral blood smear to examine RBC indices
Normocytic and normochromic erythrocytes (normal or slightly low mean corpuscular volume [MCV]).
Nursing Diagnoses and Interventions
Activity intolerance
Nursing Interventions
 Patient-Family Teaching and Discharge Planning
Patient-Family Teaching and Discharge Planning
Include verbal and written information about the following:
 Hemolytic and Sickle Cell Anemia
Hemolytic and Sickle Cell Anemia
Overview/Pathophysiology
Hemolytic anemia is characterized by abnormal or premature destruction of RBCs. Hemolysis can occur because of intrinsic or extrinsic factors (Table 8-1), for example, from a foreign antigen (e.g., from a transfusion reaction) or an autoimmune reaction in which the hemolytic agent is intrinsic to the patient’s body. Other possible causes include exposure to radiation and ingestion of certain medications (e.g., sulfisoxazole, phenytoin, methyldopa). Acquired hemolytic anemia is usually the result of an abnormal immune response that causes premature destruction of RBCs.
Assessment
Signs and symptoms/physical findings
Acute indicators (hemolytic crisis)
Fever; headache; visual blurring or temporary blindness; severe abdominal pain; vomiting; splenomegaly; hepatomegaly; back, lower leg, and joint pain; priapism; palpitations; shortness of breath (because of pulmonary sequestration, anemia, infection); aplastic crisis (resulting from transient marrow suppression by viruses); chills; lymphadenopathy; decreased urinary output; and stroke. Peripheral nerve damage can result in paralysis or paresthesias. Occasionally a low-grade fever may occur 1-2 days after a crisis event. Attacks last a few hours to a few days and resolve spontaneously.
Collaborative Management
Erythrocytapheresis (RBC exchange or partial exchange)
A procedure that removes abnormal RBCs and infuses healthy RBCs with or without normal saline to correct the anemia. It is used for younger patients with a history of stroke or for individuals with pulmonary or cardiac disease.
Stem cell transplant from human leukocyte antigen (HLA)–matched donor following high-dose chemotherapy
Nursing Diagnoses and Interventions
Chronic pain
related to joint hemolysis secondary to hemolytic crisis or sickle cell disease
Nursing Interventions
Ineffective peripheral and cardiopulmonary tissue perfusion
related to inflammatory process and occlusion of blood vessels with RBCs
Desired outcome
Following treatment, patient has adequate peripheral and cardiopulmonary perfusion, as evidenced by SBP 10 mm Hg or less lower than baseline SBP, peripheral pulses 2+ or more on a 0-4+ scale, heart rate (HR) 100 beats per minute (bpm) or less, respiratory rate (RR) 12-20 breaths/min with normal depth and pattern (eupnea), and normal skin color.
Nursing Interventions
Nursing Interventions
 Patient-Family Teaching and Discharge Planning
Patient-Family Teaching and Discharge Planning
Include verbal and written information about the following:
 Aplastic Anemia
Aplastic Anemia


