Chapter 3 Genetic Health Breakdown
When you have completed this chapter you will be able to
INTRODUCTION
Unless we have an identical twin, we are all genetically different, and the study of human genetics involves looking at these individual differences or variations. Medical genetics applies these principles to the practice of medicine – for example, the fact that children resemble parents, and that certain diseases tend to run in families, has been known for centuries. However, the reasons for these have only become apparent over the past 150 years, and the clinical application of this knowledge is an even more recent development1.
The many complex events that result in the birth of a new baby usually occur without mishap, and the baby is ‘normal’ and adjusts successfully to life outside the uterus. Occasionally, a baby is not ‘normal’, and one of the reasons for this could be a genetic or chromosomal problem (e.g., cystic fibrosis or Down syndrome) and the parents may request advice from a clinical genetics department. However, we are not always ‘born with’ the problem; genetic health breakdown can be acquired, for example, in the case of some cancers. As a result of the Human Genome Project, many more problems that develop in adult life are known to have a genetic basis, although there is still much basic cell biology to be understood before interventions can be developed that will either prevent or treat these problems2.
This chapter commences by reviewing the assumed knowledge in the form of a quiz. Unless you understand the implications and applications of Watson and Crick’s famous discovery in 19533, you will find it difficult to support your client with the many dilemmas that genetic health breakdown can bring. As you will be aware, in this seminal work, James Watson and the late Francis Crick showed that deoxyribonucleic acid (DNA), was the genetic material, and that it was coded for hereditary characteristics.
ASSUMED KNOWLEDGE
This section reviews introductory biological science including the structure of nucleic acids and the mechanisms of DNA replication and protein synthesis and how the molecular structure of DNA enables a cell to produce an identical copy of itself (during mitosis), as in skin cells that regrow after an incision, for example. The total DNA in the nucleus of a cell, the genome, comprises many thousands of genes, and one gene contains the code to make one protein. In humans, genes are grouped together to form chromosomes. Also important is an awareness that human reproduction involves the combination of the genetic material from a sperm (spermatozoa) and an egg (oocyte), and an understanding of the sequence of events occurring from fertilisation to birth, including meiosis. Any of these processes may be involved in the specific gene or chromosome problem experienced by a client and thus, it is important to understand the structure and functioning of all of these processes. There are huge volumes of new information involving the area of medical genetics and it is essential that practitioners in the delivery of care in this area have a basic understanding of the research findings.
Complete the following after reviewing the assumed knowledge in appropriate anatomy and physiology texts. See references at the end of the chapter, for example4,5.
ASSUMED KNOWLEDGE QUESTIONNAIRE
Mark the one answer of the four that you consider the most appropriate.
(Answers to this assumed knowledge questionnaire are listed at the end of this chapter.)
MEDICAL GENETICS
As with many specialties, the vocabulary of medical genetics becomes increasingly more complex. This section will build on your previous knowledge and help you understand some of the language used by the many professionals involved in medical genetics. Sometimes, you may need to act as an ‘interpreter’ for your client, although you have probably already realised that access to the Internet has resulted in clients becoming extremely knowledgeable about many aspects of their problem, care and treatment.
WHAT ARE CHROMOSOMES AND GENES?
The human karyotype, or set of chromosomes, is made up of 46 chromosomes arranged in recognisable pairs; one member of the pair comes from the mother and the other from the father. Of the 46, 22 pairs are called autosomes, which direct many body activities, and the final pair is the sex chromosomes. If the sex chromosomes are identical in size (XX), the individual is female; if they are different (XY), the individual is male. All pairs of chromosomes can be identified based on their size, the banding pattern and the position of a constricted portion, called the centromere (see Figures 3.1 and 3.2).
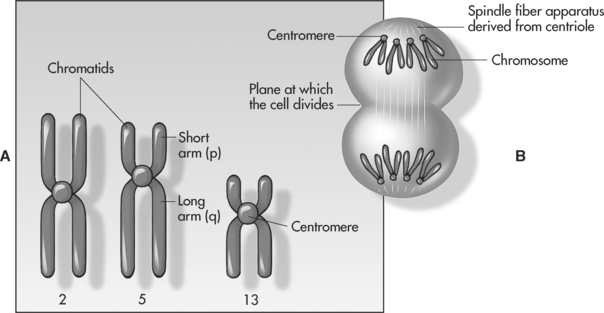
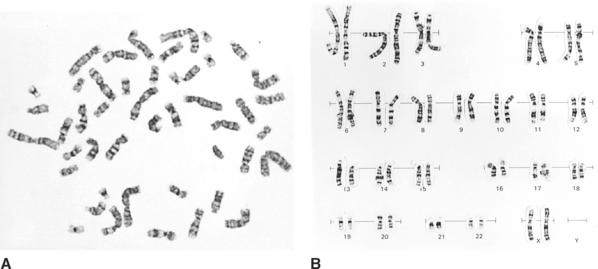
The identification and counting (‘mapping’) of all 46 chromosomes is an important cytogenetic test and is called karyotyping. A normal female and male karyotype would be reported as 46, XX or 46, XY respectively6.
Chromosomes carry our genes and each pair of autosomes is known as a homologous pair because they each code for the same features, carrying genes for the same characteristic in the same positions along the chromosome. Some of the larger chromosomes can carry more than 4500 genes4. These paired genes may be the same or different. Different genes for the same feature are called alleles of the gene, for example, tallness and shortness alleles are forms of the gene for height. One allele can be dominant to the other, which is referred to as recessive. A dominant gene is expressed, whereas recessive genes are only expressed if both copies are recessive alleles. If the alleles are identical, they are said to be homozygous; if they are different, heterozygous.
To summarise then, a gene is a DNA sequence coding for a protein or polypeptide. Our genes characterise us as individuals, one gene from each pair coming from our father and the other from our mother. The genotype is the genetic makeup of the individual and the phenotype is how a person looks or functions, based on the genotype4 (see Table 3.1).
Table 3.1: EXAMPLES OF HUMAN PHENOTYPE, DETERMINED BY SIMPLE DOMINANT-RECESSIVE INHERITANCE. (Note: Aa – a capital letter refers to the dominant allele and the small letter to a recessive allele)
| Genotype (Aa or Aa) | Genotype (aa) |
|---|---|
| Free earlobes | Attached earlobes |
| Far sightedness | Normal vision |
| Double-jointed thumbs | Normal joint flexibility |
| Normal skin pigmentation | Albinism |
CAN MISTAKES OCCUR IN CHROMOSOMES AND GENES?
Most mutations are either repaired by special ‘repair enzymes’ or the affected daughter cell will die. If an abnormal protein is produced, ‘protein destructor enzymes’ or proteases may get rid of it. For some reason, this does not always happen and these mutations can cause problems such as cancer in somatic daughter cells. If the fault occurs in germ cells, the mutation will be inherited and may lead to an inherited disease. However, there are many mutations that do not cause problems, known as neutral mutations.
SINGLE GENE MUTATIONS
Examples of single gene mutations include
VISIBLE CHROMOSOME MUTATIONS
Examples of visible chromosome mutations include
Single gene disorders commonly follow three patterns of inheritance, autosomal dominant (AD), autosomal recessive (AR) and X-linked. Generally, an AD disorder needs only one parent to have the gene fault; in an AR disorder both parents ‘carry’ the faulty gene and in an X-linked disorder, males will express the disorder and females will be carrier. These inheritance patterns will be demonstrated in the case studies. For further information, see references1,7.
WHAT ARE EXAMPLES OF MUTATIONS IN SPECIFIC PROTEINS THAT CAUSE DISEASE?
Dystrophin is a protein in the cytoplasm of muscle cells that links the inside of the cells with the outside, via proteins in the cell membrane, to provide stability to the cell membrane when the fibre contracts. Dystrophin is absent in Duchenne muscular dystrophy (DMD) and as a consequence, the cell membrane tears during contraction. Ultimately, the damage leads to the muscle fibres shrinking and dying, with the tragic consequences in affected boys. Mutations in enzymes may result in no enzyme being synthesised, a less active abnormal enzyme or a reduced amount of the normal enzyme. Enzymes are essential for cellular activities and metabolic processes, and a gene fault in an enzyme will affect the particular process catalysed by the enzyme. The consequence could be the build-up of the basic substrate (the initial ‘substance’ catalysed by the particular metabolic pathway), leading to damage in the tissues, as seen in, for example, phenylketonuria (PKU). If the gene fault in the enzyme caused a block in the metabolic process, a decreased amount of end product occurs, for example, in albinism, where a reduced amount of melanin is synthesised. Thalassaemia and sickle cell anaemia result from mutations that affect the synthesis of the polypeptide chains in haemoglobin. One of the case studies will be looking at cystic fibrosis, which is a defect in a membrane protein, involved in ion transport into the cell6.
To recap, there are genetically inherited disorders passed on from parent to child, and genetically acquired disorders that develop during life. It is also apparent that many disorders involve several genes interacting with the environment to increase the susceptibility to a disease, such as schizophrenia and breast cancer. These are known as polygenic disorders. Certain inherited disorders are also more common within specific communities, for example, Tay–Sachs disease in Ashkenazi Jewry, and thalassaemia in the Greek community6.
CAN GENE PROBLEMS BE CORRECTED?
As more has become known about the basis of gene faults, the obvious question is: Can any of these faults be corrected? From your background knowledge, you will realise the problems implicit in trying to do this; it would involve inserting the correct sequence of DNA into a cell or removing an additional sequence. How could this possibly be accomplished in gametes or somatic cells? There is much research in this area, that is, in gene therapy – the actual transfer of DNA into cells for therapeutic purposes. Advances in molecular technology have led to there being clinical trials in lung diseases (e.g., cystic fibrosis), blood disorders (e.g., haemophilia), neurological and musculoskeletal disorders, as well as cancer5.
WHAT SERVICES ARE PROVIDED WITHIN MEDICAL GENETICS?
It is apparent that clients with genetic problems need very specific support and information, and this is provided by three main areas or departments involved in medical genetics, each with their own specialist staff. Clinical genetics involves counselling, examination, diagnosis and follow-up given by specialist staff, including nurses, doctors and counsellors, to individuals, couples and families concerned about specific genetic disorders. This would include assessment of risks and an explanation of reproductive options to couples at high risk of having a child with a serious genetic disorder. Those who may have chosen not to have children because of a devastating genetic disorder, for example, the female is a carrier for Duchenne muscular dystrophy, now have the possibility of having a healthy child with screening tests, high-resolution ultrasound scanning and prenatal diagnosis (PND), which includes amniocentesis and chorionic villus sampling (CVS). In molecular genetics, scientists compare genetic material (DNA), looking for specific mutations in clients undergoing predictive or direct testing. They can also compare genetic material from parents and their fetus to see if the fetus has inherited a serious genetic disorder. If the specific gene fault is not known, they are also able to trace markers through various family members to determine risks to individuals within the family. The scientists in cytogenetics look for chromosome abnormalities, and there are now many sophisticated techniques to demonstrate these1.
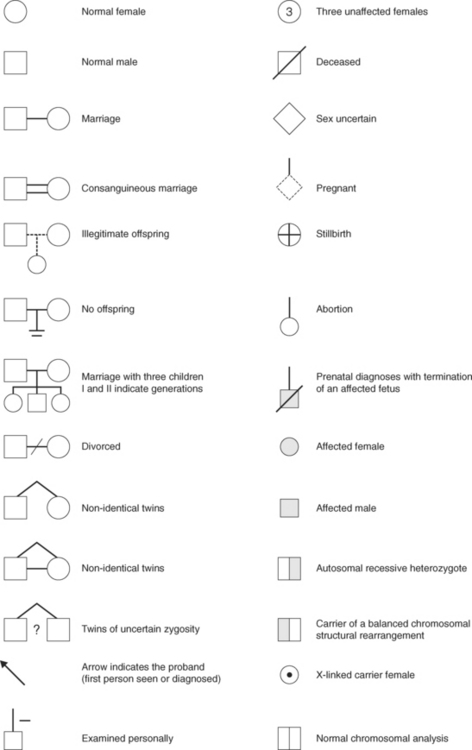
Genetic counselling is the communicating of advice about inherited conditions and an important part involves taking a detailed family history and constructing a pedigree. The symbols in Figure 3.3 are used in drawing the pedigree; you will use them when analysing the three case studies.
HUNTINGTON’S DISEASE
WHAT IS HUNTINGTON’S DISEASE?
Huntington’s disease (HD) is a rare autosomal dominant (AD) disease that is characterised by progressive movement disorders, very often deterioration in some aspects of mental health, and ultimately dementia. It gradually becomes apparent between the ages of 30 and 55 years, although the age of onset can vary from early childhood until old age. Cognitive impairment may occur before the disease is apparent8.
WHAT NEEDS TO BE REVIEWED TO UNDERSTAND HD?
An understanding of HD requires review of the following: AD inheritance; chromosomes; genes and protein synthesis; and structure and function of neurones and the areas of the brain affected – basal ganglia, cerebral cortex; neurotransmitters; normal movement; and stem cells4,5.
WHAT IS THE CAUSE (AETIOLOGY) OF HD?
The cause of HD is an abnormal gene on the short arm of chromosome 4 (4p16.3), the HD gene, which was discovered in 19939. The normal HD gene codes for a protein called huntingtin. The function of this protein is not known, but it is clearly necessary for normal brain cell function. The normal gene contains 10 to 34 copies of a CAG codon (cytosine-adenine-guanine), which codes for the amino acid glutamine. However, when the disease-causing gene is present, there are many more repeats, the tendency being that the larger the number, the earlier the age of onset of the disease. Most adult-onset clients have repeats ranging from 40 to 5010. If the abnormal HD gene is passed from mother to offspring, the number of repeats tends to be the same, but when passed from the father, there is often an increase in the number of repeats, which could lead to an earlier onset of the disease. This phenomenon is known as ‘anticipation’ and it can be seen when looking at the family tree (pedigree) that the age of onset tends to decrease in successive generations. In the juvenile form of HD, with an age of onset between infancy and 20, the repeats exceed 5510.
THE CONSEQUENCE OF HAVING THE ABNORMAL HD GENE (THE PATHOGENESIS)
Inheriting the HD gene leads to the abnormal form of the huntingtin protein. Although found in the cytoplasm of all cells of the body, huntingtin selectively kills neurones. There are several theories as to how it does this, but at the simplest level, it may be that the abnormal protein is folded differently and proteases, which would normally remove abnormal proteins, are unable to do so. The abnormal huntingtin then triggers a process leading to protein aggregation and inclusions within the nuclei, which ultimately cause the cells to die. The part of the brain most affected is the basal ganglia, which organises motor movement. The neurones in the caudate nucleus and the putamen, together known as the striatum, are the first to die. Understanding the function of the basal ganglia enables understanding of the effects of HD. The motor cortex is responsible for planning and executing body movement, and there are two pathways between the motor cortex and the basal ganglia. As cells of the striatum die, both pathways are affected. The over-stimulation of one pathway leads to irregular, jerky movements called chorea and under-stimulation of the other leads to slow motor movements. Death of other neurones occurs, especially in the cerebral cortex, affecting the limbic system controlling emotional behaviour, and other areas controlling arousal. As nerve cells die, the neurotransmitters normally secreted by the cells decrease. The neurotransmitter that is depleted most is gamma-amino butyric acid or GABA, which leads to an imbalance of other neurotransmitters6,11–13.
SUMMARY OF THE CLINICAL FEATURES
Motor symptoms often precede cognitive impairment. However, about 10 years prior to the onset of the motor disorders, mild psychotic and behavioural features may be apparent14. The movement disorders include involuntary, jerking movements of limbs, head and trunk (chorea=dance) and slow motor movements (athetosis), and significant problems with speech articulation (dysarthria). There is progressive intellectual decline; memory loss and judgment are impaired. Mental ill health includes depression, neurosis, personality changes, delusions and an increased risk of suicide15. Progressive dementia includes loss of inhibition, emotional lability, verbal and emotional outbursts leading, ultimately, to severe dementia.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


