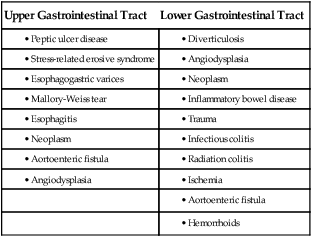
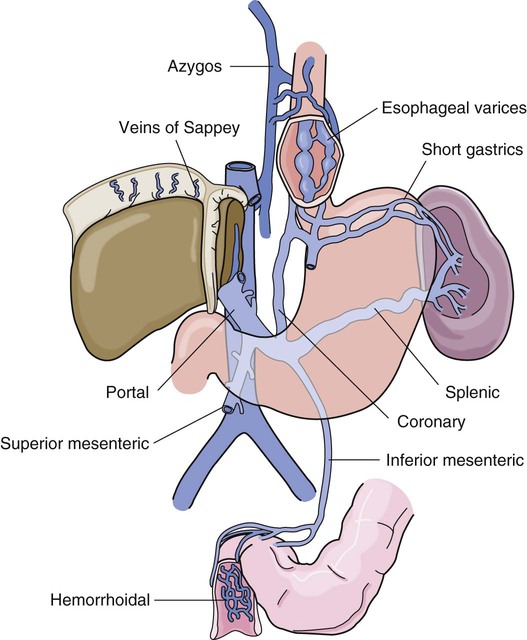
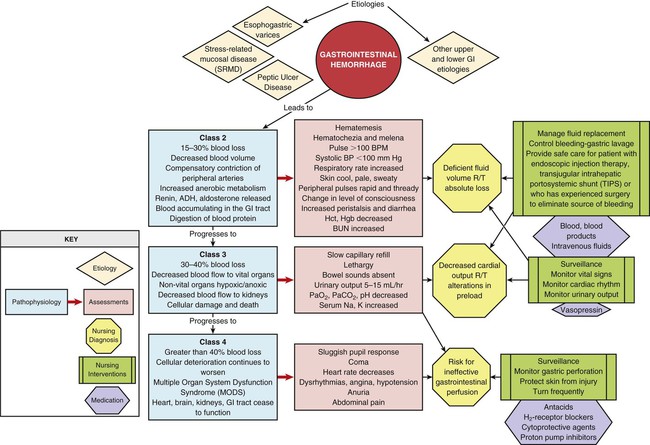
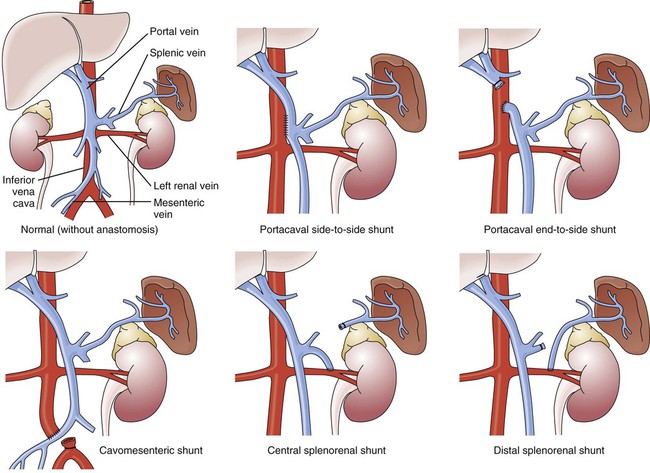
GI hemorrhage is a potentially life-threatening emergency that remains a common complication of critical illness and results in over 300,000 hospital admissions yearly.1 Despite advances in medical knowledge and nursing care, the mortality rate for patients with acute GI bleeding remains at 10% per annum in the United States.1 GI hemorrhage occurs from bleeding in the upper or lower GI tract. The ligament of Treitz is the anatomic division used to differentiate between the two areas. Bleeding proximal to the ligament is considered to be from the upper GI tract, and bleeding distal to the ligament is considered to be from the lower GI tract.1–3 The various causes of acute GI hemorrhage are listed in Box 30-1.4 Only the three main causes of GI hemorrhage commonly seen in the critical care unit are discussed further. Peptic ulcer disease (i.e., gastric and duodenal ulcers), which results from the breakdown of the gastromucosal lining, is the leading cause of upper GI hemorrhage, accounting for approximately 40% of cases.5,6 Normally, protection of the gastric mucosa from the digestive effects of gastric secretions is accomplished in several ways. First, the gastroduodenal mucosa is coated by a glycoprotein mucous barrier that protects the surface of the epithelium from hydrogen ions and other noxious substances present in the gut lumen.6–8 Adequate gastric mucosal blood flow is necessary to maintain this mucosal barrier function. Second, gastroduodenal epithelial cells are protected structurally against damage from acid and pepsin because they are connected by tight junctions that help prevent acid penetration. Third, prostaglandins and nitric oxide protect the mucosal barrier by stimulating the secretion of mucus and bicarbonate and inhibiting the secretion of acid.6–8 Peptic ulceration occurs when these protective mechanisms cease to function, allowing gastroduodenal mucosal breakdown. After the mucosal lining is penetrated, gastric secretions autodigest the layers of the stomach or duodenum, leading to injury of the mucosal and submucosal layers. This results in damaged blood vessels and subsequent hemorrhage. The two main causes of disruption of gastroduodenal mucosal resistance are the bacterial action of Helicobacter pylori and nonsteroidal anti-inflammatory drugs (NSAIDs).9 Stress-related mucosal disease (SRMD) is an acute erosive gastritis that covers both types of mucosal lesions that are often found in the critically ill patient: stress-related injury and discrete stress ulcers.10–12 Additional terms used to describe this condition include stress ulcers, stress erosions, stress gastritis, hemorrhagic gastritis, and erosive gastritis. These abnormalities develop within hours of admission.11 They range from superficial mucosal erosions to deep focal lesions and usually affect the upper GI tract.11 SRMD occurs by means of the same pathophysiologic mechanisms as peptic ulcer disease, but the main cause of disruption of gastric mucosal resistance is increased acid production and decreased mucosal blood flow, resulting in ischemia and degeneration of the mucosal lining.11 Patients at risk include those in situations of high physiologic stress, as occurs with mechanical ventilation, extensive burns, severe trauma, major surgery, shock, sepsis, coagulopathy, or acute neurologic disease.13 SRMD is decreasing in incidence because of advances in therapeutic techniques and prevention of hypoperfusion of the mucosa.14 Esophagogastric varices are engorged and distended blood vessels of the esophagus and proximal stomach that develop as a result of portal hypertension caused by hepatic cirrhosis, a chronic disease of the liver, which results in damage to the liver sinusoids (Fig. 30-1). Without adequate sinusoid function, resistance to portal blood flow is increased, and pressures within the liver are elevated. This leads to increased portal venous pressure (portal hypertension), causing collateral circulation to divert portal blood from areas of high pressure within the liver to adjacent areas of low pressure outside the liver, such as into the veins of the esophagus, the spleen, the intestines, and the stomach. The tiny, thin-walled vessels of the esophagus and proximal stomach that receive this diverted blood lack sturdy mucosal protection. The vessels become engorged and dilated, forming esophagogastric varices that are vulnerable to damage from gastric secretions and that may result in subsequent rupture and massive hemorrhage.15 The risk of variceal bleeding increases with disease severity and variceal size, but overall, bleeding occurs in 25% to 30% of patients within 2 years of diagnosis, and 20% to 30% mortality from each bleeding episode.16,17 GI hemorrhage is a life-threatening disorder that is characterized by acute, massive bleeding. Regardless of the cause, acute GI hemorrhage results in hypovolemic shock, initiation of the shock response, and development of multiple organ dysfunction syndrome if left untreated (see Concept Map, Fig. 30-2).7 However, the most common cause of death in cases of GI hemorrhage is exacerbation of the underlying disease, not intractable hypovolemic shock. The initial clinical presentation of the patient with acute GI hemorrhage is that of a patient in hypovolemic shock, and the clinical presentation depends on the amount of blood lost (Table 30-1).7 Hematemesis (bright red or brown, “coffee grounds” emesis), hematochezia (bright red stools), and melena (black, tarry, or dark red stools) are the hallmarks of GI hemorrhage.5,18. TABLE 30-1 CLINICAL CLASSIFICATION OF HEMORRHAGE From Klein DG. Physiologic response to traumatic shock. AACN Clin Issues Crit Care Nurs. 1990;1:505. The patient who is vomiting blood is usually bleeding from a source above the duodenojejunal junction; reverse peristalsis is seldom sufficient to cause hematemesis if the bleeding point is below this area. The hematemesis may be bright red or look like coffee grounds, depending on the amount of gastric contents at the time of bleeding and the length of time the blood has been in contact with gastric secretions. Gastric acid converts bright red hemoglobin to brown hematin, accounting for the coffee grounds appearance of the emesis. Bright red emesis results from profuse bleeding with little contact with gastric secretions.19 Laboratory tests can help determine the extent of bleeding, although the patient’s hemoglobin level and hematocrit are poor indicators of the severity of blood loss if the bleeding is acute. As whole blood is lost, plasma and red blood cells are lost in the same proportion; if the patient’s hematocrit is 45% before a bleeding episode, it will be 45% several hours later.7 It may take 24 to 72 hours for the redistribution of plasma from the extravascular space to the intravascular space to occur and cause the patient’s hemoglobin level and hematocrit value to decrease.19 To isolate and treat the source of bleeding, an urgent fiberoptic endoscopy is usually undertaken.20 Before endoscopy, the patient must be hemodynamically stabilized.21 Tagged red blood cell scanning, angiography, or both, may be done to assist with localizing and treating a bleeding lesion in the GI tract when it is impossible to clearly view the GI tract because of continued active bleeding.19 To reduce mortality related to GI hemorrhage, patients at risk should be identified early, and interventions should be implemented to reduce gastric acidity and support the gastric mucosal defense mechanisms. Management of the patient at risk for GI hemorrhage should include prophylactic administration of pharmacologic agents for neutralization of gastric acids. These agents include antacids, histamine-2 (H2) antagonists, cytoprotective agents, and proton pump inhibitors (PPIs).5,22 Priorities in the medical management of the patient with GI hemorrhage include airway protection, fluid resuscitation to achieve hemodynamic stability, correction of co-morbid conditions (e.g., coagulopathy), therapeutic procedures to control or stop bleeding, and diagnostic procedures to determine the exact cause of the bleeding.5,21 The initial treatment priority is the restoration of adequate circulating blood volume to treat or prevent shock. This is accomplished with the administration of intravenous infusions of crystalloids, blood, and blood products.22 Hemodynamic monitoring can help guide fluid replacement therapy, particularly in patients at risk for heart failure.7 Supplemental oxygen therapy is initiated to increase oxygen delivery and improve tissue perfusion.7,22 A large nasogastric tube may be inserted to confirm the diagnosis of active bleeding, to facilitate gastric lavage, decrease the risk for aspiration and to prepare the esophagus, stomach, and proximal duodenum for endoscopic evaluation.5 Interventions to control bleeding are the second priority for the patient with GI hemorrhage. In the patient with GI hemorrhage related to peptic ulcer disease, bleeding hemostasis may be accomplished by endoscopic injection therapy in conjunction with thermal or hemostatic clips.20 Endoscopic thermal therapy uses heat to cauterize the bleeding vessel, and endoscopic injection therapy uses a variety of agents such as hypertonic saline, epinephrine, ethanol, and sclerosants to induce localized vasoconstriction of the bleeding vessel.6 Intra-arterial infusion of vasopressin into the gastric artery or intra-arterial injection of an embolizing agent (e.g., Gelfoam pledgets, polyvinyl alcohol particles, coils) can be performed during arteriography to control bleeding after the site has been identified.19 In acute variceal hemorrhage, control of bleeding may be initially accomplished through the use of pharmacologic agents and endoscopic therapies. Intravenous vasopressin, somatostatin, and octreotide can reduce portal venous pressure and slow variceal hemorrhaging by constricting the splanchnic arteriolar bed.16 Two commonly used endoscopic therapies are endoscopic injection sclerotherapy (EIS) and endoscopic variceal ligation (EVL).23 EIS controls bleeding by the injection of a sclerosing agent in or around the varices. This creates an inflammatory reaction that induces vasoconstriction and results in the formation of a venous thrombosis. During EVL, bands are placed around the varices to create an obstruction to stop the bleeding.24 If these initial therapies fail, transjugular intrahepatic portosystemic shunting (TIPS) may be necessary. In a TIPS procedure, a channel between the systemic and portal venous systems is created to redirect portal blood, thereby reducing portal hypertension and decompressing the varices to control bleeding (Fig. 30-3).23,24 The patient who remains hemodynamically unstable despite volume replacement may need urgent surgery. Surgical intervention is required to control bleeding in a minority of patients.25 The operative procedure of choice to control bleeding from peptic ulcer disease is a vagotomy and pyloroplasty. During this procedure, the vagus nerve to the stomach is severed, eliminating the autonomic stimulus to the gastric cells and reducing hydrochloric acid production. Because the vagus nerve also stimulates motility, a pyloroplasty is performed to provide for gastric emptying.26 If medical treatment is unsuccessful and angiographic interventional TIPS procedure is not available, operative procedures to control bleeding gastroesophageal varices may be undertaken. Though rarely performed, operative interventions focus on some form of shunting (Fig 30-4).25 These shunt procedures are also referred to as decompression procedures because they result in the diversion of portal blood flow away from the liver and decompression of the portal system. The portacaval shunt procedure has two variations: (1) an end-to-side portacaval shunt procedure, which involves the ligation of the hepatic end of the portal vein with subsequent anastomosis to the vena cava; and (2) a side-to-side portacaval shunt procedure, during which the side of the portal vein is anastomosed to the side of the vena cava. A mesocaval shunt procedure involves the insertion of a graft between the superior mesenteric artery and the vena cava. During a distal splenorenal shunt procedure, the splenic vein is detached from the portal vein and anastomosed to the left renal vein.27 All critically ill patients should be considered at risk for stress ulcers and, therefore, GI hemorrhage. Routine assessment of gastric fluid pH monitoring is controversial.12,28 Maintaining the pH between 3.5 and 4.5 is a goal of prophylactic therapy. Gastric pH measurements made with litmus paper or direct nasogastric tube probes may be used to assess gastric fluid pH and the effectiveness or need for prophylactic agents.28 Patients at risk also should be assessed for the presence of bright red or coffee grounds emesis, bloody nasogastric aspirate, and bright red, black, or dark red stools.4 Any signs of bleeding should be promptly reported to the physician. Nursing management of a patient experiencing acute GI hemorrhage incorporates a variety of nursing diagnoses (Box 30-2). Nursing interventions include administering volume replacement, controlling the bleeding, providing comfort and emotional support, maintaining surveillance for complications, and educating the patient and family. Acute Gastrointestinal Hemorrhage • Deficient Fluid Volume, related to absolute loss, p. 1132 • Decreased Cardiac Output, related to alterations in preload, p. 1128 • Risk for Aspiration, p. 1154 • Imbalanced Nutrition: Less Than Body Requirements, related to lack of exogenous nutrients and increased metabolic demand, p. 1143 • Powerlessness, related to health care environment or illness-related regimen, p. 1152 • Compromised Family Coping, related to critically ill family member, p. 1127 • Deficient Knowledge, related to lack of previous exposure to information, p. 1134 (see Patient Education, Box 30-3) Measures to facilitate volume replacement include obtaining intravenous access and administering prescribed fluids and blood products. Two large-diameter peripheral intravenous catheters should be inserted to facilitate the rapid administration of prescribed fluids.1 One measure to control active bleeding is gastric lavage. It is used to decrease gastric mucosal blood flow and evacuate blood from the stomach. Gastric lavage is performed by inserting a large-bore nasogastric tube into the stomach and irrigating it with normal saline or water until the returned solution is clear. It is important to keep accurate records of the amount of fluid instilled and aspirated to ascertain the true amount of bleeding.1 Historically, iced saline was favored as a lavage irrigant. Research has shown, however, that low-temperature fluids shift the oxyhemoglobin dissociation curve to the left, decrease oxygen delivery to vital organs, and prolong bleeding time and prothrombin time. Iced saline also may further aggravate bleeding; therefore, room-temperature water or saline is the preferred irrigant for use in gastric lavage.29 The patient should be continuously observed for signs of gastric perforation. Although a rare complication, gastric perforation constitutes a surgical emergency. Signs and symptoms include sudden, severe, generalized abdominal pain with significant rebound tenderness and rigidity. Perforation should be suspected when fever, leukocytosis, and tachycardia persist despite adequate volume replacement.30 Early in the hospital stay, the patient and family should be taught about acute GI hemorrhage and its causes and treatments. As the patient moves toward discharge, teaching should focus on the interventions necessary for preventing the recurrence of the precipitating disorder. If an alcohol abuser, the patient should be encouraged to stop drinking and be referred to an alcohol cessation program (Box 30-3). Collaborative management of the patient with acute GI hemorrhage is outlined in Box 30-4. Acute pancreatitis is an inflammation of the pancreas that produces exocrine and endocrine dysfunction that may also involve surrounding tissues, remote organ systems, or both. The clinical course can range from a mild, self-limiting disease to a systemic process characterized by organ failure, sepsis, and death. In approximately 80% of patients, it takes the milder form of edematous interstitial pancreatitis, whereas the other 20% develop severe acute necrotizing pancreatitis.31 Reported mortality rates for acute pancreatitis range from 2% to 15% overall, with a steadily increasing rate of about 20,000 deaths per year in the United States.32,33 Several prognostic scoring systems have been developed to predict the severity of acute pancreatitis. One of the most commonly used is Ranson criteria34 (Box 30-5). If the patient has 0 to 2 factors present, the predicted mortality rate is 2%; with 3 to 4 factors, the rate is 15%; with 5 to 6 factors, the rate is 40%; and with 7 to 8 factors, predicted mortality rate is 100%.34–36 The two most common causes of acute pancreatitis are gallstone migration and alcoholism.36 Together, they account for approximately 80% of cases.19 Less common causes are quite diverse and include surgical trauma, hypercalcemia, various toxins, ischemia, infections, and the use of certain medications (Box 30-6). In up to 20% of patients with acute pancreatitis, no etiologic factor can be determined.19,31 In acute pancreatitis, the normally inactive digestive enzymes become prematurely activated within the pancreas itself, leading to autodigestion of pancreatic tissue. The enzymes become activated through various mechanisms, including obstruction of or damage to the pancreatic duct system, alterations in the secretory processes of the acinar cells, infection, ischemia, and other unknown factors.7,36 Trypsin is the enzyme that becomes activated first. It initiates the autodigestion process by triggering the secretion of proteolytic enzymes such as kallikrein, chymotrypsin, elastase, phospholipase A, and lipase. Release of kallikrein and chymotrypsin results in increased capillary membrane permeability, leading to leakage of fluid into the interstitium and the development of edema and relative hypovolemia. Elastase is the most harmful enzyme in terms of direct cell damage. It dissolves the elastic fibers of blood vessels and ducts, leading to hemorrhage. Phospholipase A, in the presence of bile, destroys the phospholipids of cell membranes, causing severe pancreatic and adipose tissue necrosis. Lipase flows into the damaged tissue and is absorbed into the systemic circulation, resulting in fat necrosis of the pancreas and surrounding tissues.7,37 The extent of injury to the pancreatic cells determines the type of acute pancreatitis that develops. If injury to the pancreatic cells is mild and without necrosis, edematous pancreatitis develops. The acinar cells appear structurally intact, and blood flow is maintained through small capillaries and venules. This form of acute pancreatitis is self-limiting. If injury to the pancreatic cells is severe, acute necrotizing pancreatitis develops.32,37 Cellular destruction in pancreatic injury results in the release of toxic enzymes and inflammatory mediators into the systemic circulation and causes injury to vessels and other organs distant from the pancreas; this may result in systemic inflammatory response syndrome (SIRS), multiorgan failure, and death.19,36 Local tissue injury results in infection, abscess and pseudocyst formation, disruption of the pancreatic duct, and severe hemorrhage and shock.19 The clinical manifestations of acute pancreatitis range from mild to severe and often mimic those of other disorders (Box 30-7). Acute onset of abdominal pain, nausea, and vomiting are hallmark symptoms.19,32,37 Epigastric to periumbilical pain may vary from mild and tolerable to severe and incapacitating. Many patients report a twisting or knifelike sensation that radiates to the low dorsal region of the back. The patient may obtain some comfort by leaning forward or assuming a semifetal position. Other clinical findings include fever, diaphoresis, weakness, tachypnea, hypotension, and tachycardia. Depending on the extent of fluid loss and hemorrhage, the patient may exhibit signs of hypovolemic shock.19,37 The results of physical assessment usually reveal hypoactive bowel sounds and abdominal tenderness, guarding, distention, and tympany. Findings that may indicate pancreatic hemorrhage include Grey Turner sign (gray-blue discoloration of the flanks) and Cullen sign (discoloration of the umbilical region); however, they are rare and usually seen several days into the illness.19 A palpable abdominal mass indicates the presence of a pseudocyst or abscess.19 Assessment of laboratory data usually demonstrates elevated levels of serum amylase and lipase. Serum lipase is more pancreas specific than amylase and a more accurate marker for acute pancreatitis. Amylase is present in other body tissues, and other disorders (e.g., intra-abdominal emergencies, renal insufficiency, salivary gland trauma, liver disease) may contribute to an elevated level. Unlike other serum enzymes, however, amylase is excreted in urine, and this clearance increases with acute pancreatitis. Measurement of urinary versus serum amylase should be considered in light of the patient’s creatinine clearance. The serum amylase level may be elevated for only 3 to 5 days; if the patient delays seeking treatment, a normal level (false-negative result) may be detected. A marker of severity may be determined with a serum cross-reactive (C-reactive) protein level.19,38 Leukocytosis, hypocalcemia, hyperglycemia, hyperbilirubinemia, and hypoalbuminemia may also be present (Table 30-2).19,36,38 TABLE 30-2 LABORATORY TESTS AND DIAGNOSTIC PROCEDURES FOR ACUTE PANCREATITIS Modified from Krumberger JM. Acute pancreatitis. Crit Care Nurs Clin North Am. 1993;5:185. An abdominal ultrasound scan is obtained as part of the diagnostic evaluation to determine the presence of biliary stones. Contrast-enhanced computed tomography (CT) is considered the gold standard for diagnosing pancreatitis and for ascertaining the overall degree of pancreatic inflammation and necrosis.19,32 Initial management of the patient with severe acute pancreatitis includes ensuring adequate fluid and electrolyte replacement, providing nutritional support, and correcting metabolic alterations.35 Careful monitoring for systemic and local complications is critical. Because pancreatitis if often associated with massive fluid shifts, intravenous crystalloids and colloids are administered immediately to prevent hypovolemic shock and maintain hemodynamic stability. Electrolytes are monitored closely, and abnormalities such as hypocalcemia, hypokalemia, and hypomagnesemia are corrected.37 If hyperglycemia develops, exogenous insulin may be required. Over the past three decades, nutritional support has shifted. Previously, conventional nutritional management was to place the patient on a nothing-by-mouth (NPO) regimen and institute intravenous hydration. The rationale was to rest the inflamed pancreas and prevent enzyme release. Enteral or parental support should be initiated if oral intake is withheld more than 5 to 7 days.39 Randomized clinical trials have demonstrated that enteral feeding (gastric or jejunal) is safe and cost effective and that it is associated with fewer septic and metabolic complications than other methods.40–42 Enteral feeding enhances immune modulation and maintenance of the intestinal barrier, and it avoids complications associated with parental nutrition. Early initiation of enteral feeding is preferred over TPN.39 However, TPN still has a role for the critically ill patient with acute pancreatitis who does not tolerate enteral feeding or when nutritional goals are not reached within 2 days.37,41,42 In the past, nasogastric suction was also recommended, but this intervention has not been shown to be beneficial and should be instituted only if the patient has persistent vomiting, obstruction, or gastric distention.37
Gastrointestinal Disorders and Therapeutic Management
Acute Gastrointestinal Hemorrhage
Description
Etiology
Peptic Ulcer Disease
Stress-Related Mucosal Disease
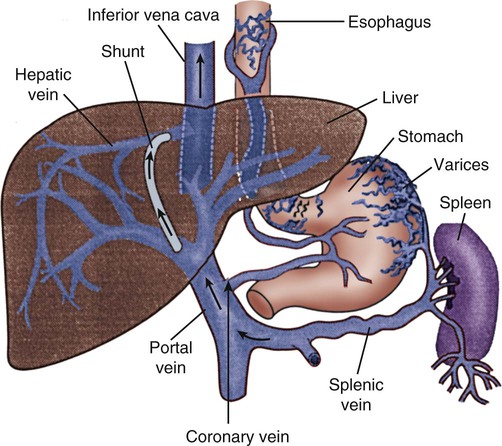
Esophagogastric Varices
Pathophysiology
Assessment and Diagnosis
CLASS
BLOOD LOSS (%)
CLINICAL SIGNS AND SYMPTOMS
1
≤15
Pulse rate: normal or <100 beats/min (supine)
Capillary refill <3 seconds
Urine output: adequate (30-35 mL/hr)
Orthostatic hypotension
Apprehensive
2
15-30
Pulse rate: increased (>100 beats/min)
Capillary refill: sluggish
Pulse pressure: decreased
Blood pressure: normal (supine)
Tachypnea
Urine output: low (25-30 mL/hr)
3
30-40
Pulse rate: 120+ beats/min (supine)
Hypotension
Skin: cool, pale
Confused
Hyperventilating
Urine output: low (5-15 mL/hr)
4
≥40
Profoundly hypotensive
Pulse rate: 140+ beats/min
Confused, lethargic
Urine output minimal
Hematemesis
Laboratory Studies
Diagnostic Procedures
Medical Management
Stabilization
Controlling the Bleeding
Peptic Ulcer Disease.
Esophagogastric Varices.
Surgical Intervention
Peptic Ulcer Disease.
Esophagogastric Varices.
Nursing Management
 Box 30-2 Nursing Diagnoses
Box 30-2 Nursing Diagnoses
Administering Volume Replacement.
Controlling the Bleeding.
Maintaining Surveillance for Complications.
Educating the Patient and Family.
Acute Pancreatitis
Description
Etiology
Pathophysiology
Assessment and Diagnosis
Physical Examination
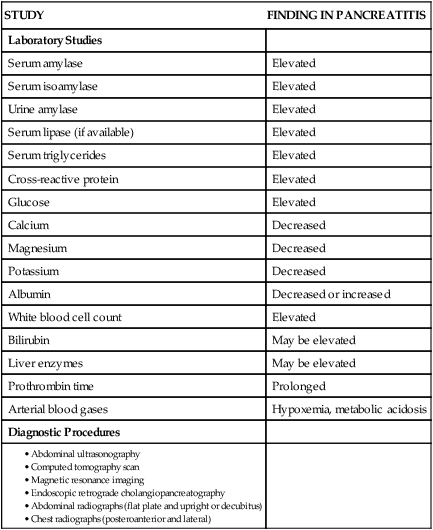
Laboratory Studies
STUDY
FINDING IN PANCREATITIS
Laboratory Studies
Serum amylase
Elevated
Serum isoamylase
Elevated
Urine amylase
Elevated
Serum lipase (if available)
Elevated
Serum triglycerides
Elevated
Cross-reactive protein
Elevated
Glucose
Elevated
Calcium
Decreased
Magnesium
Decreased
Potassium
Decreased
Albumin
Decreased or increased
White blood cell count
Elevated
Bilirubin
May be elevated
Liver enzymes
May be elevated
Prothrombin time
Prolonged
Arterial blood gases
Hypoxemia, metabolic acidosis
Diagnostic Procedures
Diagnostic Procedures
Medical Management
Fluid Management
Nutritional Support
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Gastrointestinal Disorders and Therapeutic Management
Get Clinical Tree app for offline access
