Bruises, cuts, or scrapes or uncomplicated joint hemorrhage
Nose, mouth, and gum bleeds; dental extractions; hematuria
Joint or muscle hemorrhage, major trauma, hematuria, intracranial and/or intraperitoneal bleeding

IU, International unit.
| Alphanate Dose Guidelines for Prophylaxis During Surgery and Invasive Procedures of Adult and Pediatric Patients with von Willebrand Disease (Except Type 3 Patients Undergoing Surgery) | |
| Bleeding Prophylaxis for Surgical or Invasive Procedures | Dosage (AHF vWF:RCof IU/kg body weight) |
| Adult | Preoperative dose: 60 vWF:RCof IU/kg body weight. Subsequent infusions: 40 to 60 vWF:RCof IU/kg body weight at 8- to 12-hour intervals as clinically needed. Dosing may be reduced after the third postoperative day. Continue treatment until healing is complete. |
| Adult | Minor procedure: vWF activity of 40% to 50% for at least 1 to 3 days postoperatively. |
| Adult | Major procedure: vWF activity of 40% to 50% for at least 3 to 7 days postoperatively. |
| Pediatric | Initial dose: 75 vWF:RCof IU/kg body weight. Subsequent infusions: 50 to 75 vWF:RCof IU/kg body weight at 8- to 12-hour intervals as clinically needed. Dosing may be reduced after the third postoperative day. Continue treatment until healing is complete. |

IU, International unit.
Humate-P
Treatment of hemophilia A (adults):
As a general rule, 1 IU of factor VIII activity per kg body weight will increase the circulating factor VIII level by approximately 2 IU/dL. Assess adequacy of treatment by clinical effects and monitoring of factor VIII activity. See Precautions. The following general dosages are recommended for adult patients:
| Humate-P Dose Recommendations for the Treatment of Hemophilia A* | |
| Hemorrhage Event | Dosage (IU† FVIII:C/kg body weight) |
| Minor | Loading dose 15 IU FVIII:C/kg to achieve FVIII:C plasma level of approximately 30% of normal; one infusion may be sufficient If needed, half of the loading dose may be given once or twice daily for 1-2 days |
| Moderate | Loading dose 25 IU FVIII:C/kg to achieve FVIII:C plasma level of approximately 50% of normal Followed by 15 IU FVIII:C/kg every 8-12 hours for first 1-2 days to maintain FVIII:C plasma level at 30% of normal Then same dose once or twice a day for up to 7 days or until adequate wound healing |
| Life-threatening | Initially 40-50 IU FVIII:C/kg Followed by 20-25 IU FVIII:C/kg every 8 hours to maintain FVIII:C plasma level at 80%-100% of normal for 7 days Then continue the same dose once or twice a day for another 7 days to maintain the FVIII:C level at 30%-50% of normal |

*In all cases, the dose should be adjusted individually by clinical judgment of the potential for compromise of a vital structure and by frequent monitoring of factor VIII activity in the patient’s plasma.
Treatment of von Willebrand disease (vWD) (adults and pediatric patients):
As a rule, 40-80 IU vWF:RCof (corresponding to 16 to 32 IU factor VIII in Humate-P) per kg body weight given every 8 to 12 hours. Repeat doses are administered as needed based on monitoring of appropriate clinical and laboratory measures. Expected levels of vWF:RCof are based on an expected in vivo recovery of 1.5 IU/dL rise per IU/kg vWF:RCof administered. The administration of 1 IU of factor VIII per kg body weight can be expected to lead to a rise in circulating vWF:RCof of approximately 3.5 to 4 IU/dL. The following general dosages are recommended for adult and pediatric patients:
| Humate-P Dose Recommendations for Treatment of von Willebrand Disease in Adult and Pediatric Patients | ||
| Classification | Hemorrhage | Dosage (IU* vWF:RCof/kg body weight) |
| TYPE 1 | ||
| Mild (Where use of desmopressin is known or suspected to be inadequate) Baseline vWF:RCof activity typically >30% of normal (i.e., >30 IU/dL) | Major (examples) | Loading dose 40 to 60 IU/kg Then 40 to 50 IU/kg every 8 to 12 hours for 3 days to keep the nadir level of vWF:RCof >50% of normal (i.e., >50 IU/dL) Then 40 to 50 IU/kg daily for a total of up to 7 days of treatment |
| Moderate or Severe Baseline vWF:RCof activity typically <30% of normal (i.e., <30 IU/dL) | Minor (examples) | 40 to 50 IU/kg (1 or 2 doses) |
| Major (examples) | Loading dose of 50 to 75 IU/kg Then 40 to 60 IU/kg every 8 to 12 hours for 3 days to keep the nadir level of vWF:RCof >50% of normal (i.e., >50 IU/dL) Then 40 to 60 IU/kg daily for a total of up to 7 days of treatment Factor VIII:C levels should be monitored and maintained according to the guidelines for hemophilia A therapy† | |
| TYPE 2 (ALL VARIANTS) AND TYPE 3 | ||
| Minor (clinical indications above) | 40 to 50 IU/kg (1 or 2 doses) | |
| Major (clinical indications above) | Loading dose of 60 to 80 IU/kg Then 40 to 60 IU/kg every 8 to 12 hours for 3 days to keep the nadir level of vWF:RCof >50% of normal (i.e., >50 IU/dL) Then 40 to 60 IU/kg daily for a total of up to 7 days of treatment Factor VIII:C levels should be monitored and maintained according to the guidelines for hemophilia A therapy† | |
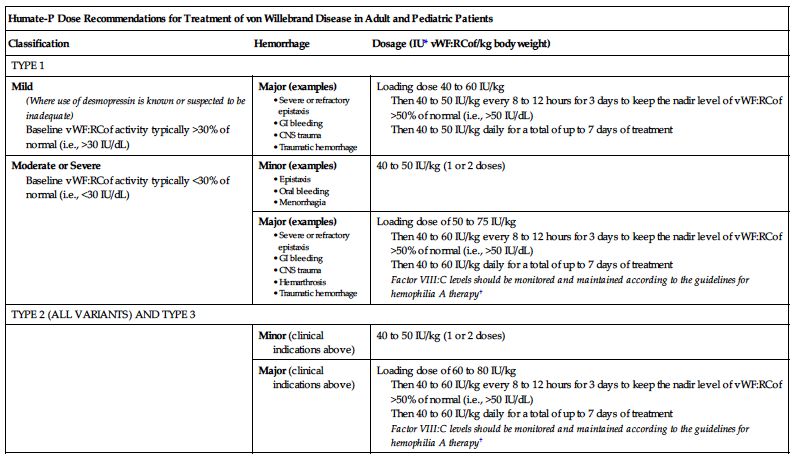
†In instances where both FVIII and vWF levels must be monitored.
Prevention of excessive bleeding during and after surgery in vWD:
In the case of emergency surgery, administer a loading dose of 50 to 60 IU/kg Humate-P and closely monitor the patient’s trough coagulation factor levels. Measurement of incremental in vivo recovery (IVR) and assessment of baseline plasma vWF:RCof and FVIII:C levels are recommended in all patients before surgery. Calculation of the loading dose requires four values: the target peak plasma vWF:RCof level, the baseline vWF:RCof level, body weight (BW) in kilograms, and IVR. If individual recovery values are not available, a standardized loading dose can be used based on an assumed vWF:RCof IVR of 2 IU/dL per IU/kg of vWF:RCof product administered.
| vWF:RCof and FVIII:C Humate-P Loading Dose Recommendations for the Prevention of Excessive Bleeding During and After Surgery | |||
| Type of Surgery | vWF:RCof Target Peak Plasma Level | FVIII:C Target Peak Plasma Level | Calculation of Loading Dose (to be administered 1 to 2 hours before surgery) |
| Major | 100 IU/dL | 80 to 100 IU/dL | Δ* vWF:RCof × BW (kg)/IVR† = IU vWF:RCof required If incremental IVR is not available, assume an IVR of 2 IU/dL per IU/kg and calculate the loading dose as follows: (100 − Baseline plasma vWF:RCof) × BW (kg)/2 In the case of emergency surgery, administer a dose of 50 to 60 IU/kg. |
| Minor/oral ‡ | 50 to 60 IU/dL | 40 to 50 IU/dL | Δ* vWF:RCof × BW (kg)/IVR† = IU vWF:RCof required |

*Δ = Target peak plasma vWF:RCof − Baseline plasma vWF:RCof.
†IVR = Incremental recovery as measured in the patient.
‡Oral surgery is defined as removal of fewer than three teeth, if the teeth are nonmolars and have no bony involvement. Removal of more than one impacted wisdom tooth is considered major surgery due to the expected difficulty of the surgery and the expected blood loss, particularly in subjects with type 2A or type 3 vWD. Removal of more than two teeth is considered major surgery in all patients.
| vWF:RCof and FVIII:C Target Trough Plasma Level and Minimum Duration of Treatment Recommendations for Subsequent Maintenance Doses of Humate-P for the Prevention of Excessive Bleeding During and After Surgery | |||||
| Type of Surgery | vWF:RCof Target Trough Plasma Levels* | FVIII:C Target Trough Plasma Levels* | Minimum Duration of Treatment | ||
| Up to 3 Days Following Surgery | After Day 3 | Up to 3 Days Following Surgery | After Day 3 | ||
| Major | >50 IU/dL | >30 IU/dL | >50 IU/dL | >30 IU/dL | 72 hours |
| Minor | ≥30 IU/dL | — | — | >30 IU/dL | 48 hours |
| Oral† | ≥30 IU/dL | — | — | >30 IU/dL | 8 to 12 hours‡ |

*Trough levels for either coagulation factor should not exceed 100 IU/dL.
†See note on oral surgery in previous chart.
‡At least one maintenance dose following surgery based on individual pharmacokinetic values.
Pediatric dose
Treatment of hemophilia A (unlabeled):
For immediate control of bleeding, follow the general recommendations for dosing and administration for adults. See Usual Dose and Maternal/Child.
Dose adjustments
Adjust subsequent doses based on FVIII:C plasma level achieved or as outlined in specific charts.
Dilution
Consult individual product instructions in the package insert; each product has a specific process for dilution. Information may be updated frequently. Alphanate provides diluent, a double-ended transfer needle, and a microaggregate filter for use in administration. Humate-P provides diluent and a filter transfer set.
Alphanate and Humate-P:
Actual number of AHF units is shown on each vial. Use only the diluent provided and maintain strict aseptic technique. Use a plastic syringe to prevent binding to glass surfaces. Warm to room temperature (25° C) before dilution and maintain throughout administration to avoid precipitation of active ingredients.
Filters:
Filters supplied by manufacturer; see Dilution.
Storage:
Alphanate: Refrigerate before use. Avoid freezing. May be stored at CRT for up to 2 months. Label vial with date removed from refrigeration. Humate-P: Store up to 25° C (up to 77° F). Avoid freezing. Alphanate and Humate-P: Do not refrigerate after reconstitution. Confirm expiration date on vial. Administer within 3 hours of reconstitution to ensure sterility. Discard any unused solution.
Compatibility
Specific information not available. Administration through a separate line without mixing with other IV fluids or medications is generally recommended for these products.
Rate of administration
Inject solution slowly. Rapid administration may result in vasomotor reactions.
Humate-P recommends a maximum rate of 4 mL/min.
Alphanate recommends a maximum rate not to exceed 10 mL/min.
Actions
A purified, sterile, lyophilized concentrate of antihemophilic factor (factor VIII) and von Willebrand Factor (vWF). Factor VIII is an essential cofactor in the activation of factor X, leading ultimately to the formation of thrombin and fibrin. It is the specific clotting factor deficient in patients with hemophilia A (classic hemophilia). vWF is important for correcting the coagulation defect in patients with von Willebrand disease (vWD). It promotes platelet aggregation and platelet adhesion on damaged vascular endothelium and acts as a stabilizing carrier protein for the procoagulant protein factor VIII. vWF activity is measured with an assay that uses an agglutinating cofactor called Ristocetin (RCof). The vWF:RCof assay provides a quantitative measurement of vWF function by determining how well vWF helps platelets adhere to one another. Reduced vWF:RCof activity indicates a deficiency of vWF. Following administration of FVIII/vWF, there is a rapid increase of plasma factor VIII activity, followed by a rapid decrease in activity and then a slower rate of decrease in activity. The mean initial half-life in hemophilic patients is 8.3 to 27.5 hours with Alphanate and 12.2 hours (range: 8.4 to 17.4 hours) with Humate-P. In patients with vWD, bleeding time decreases. Antihemophilic factor/von Willebrand Factor Complex is obtained from pooled human fresh-frozen plasma. Multiple methods of purification are used to inactivate infectious agents, including viruses.
Indications and uses
Alphanate
Prevention and control of bleeding in patients with factor VIII deficiency due to hemophilia A or acquired factor VIII deficiency. ■ Prophylaxis for surgical and/or invasive procedures in adult and pediatric patients with von Willebrand disease (vWD) (type 1 or 2) in which the use of desmopressin is either ineffective or contraindicated. ■ Not indicated for patients with severe vWD (type 3).
Humate-P
Treatment and prevention of bleeding in adult patients with hemophilia A. ■ Treatment of spontaneous and trauma-induced bleeding episodes and prevention of excessive bleeding during and after surgery in adult and pediatric patients with severe vWD or with mild or moderate vWD in which the use of desmopressin is known or suspected to be inadequate. ■ Safety and efficacy of prophylactic dosing to prevent spontaneous bleeding and to prevent excessive bleeding related to surgery have not been established in patients with vWD.
Contraindications
Alphanate:
None known when used as indicated.
Humate-P:
History of anaphylactic or severe systemic response to AHF-vWF preparations or known hypersensitivity to any of its components.
Precautions
Alphanate and Humate-P:
For IV use only. ■ Health care professionals should use caution during administration; may have risk of exposure to viral infection. ■ Important to establish that coagulation disorder is caused by factor VIII or vWF deficiency. Not useful in treatment of other deficiencies. ■ Manufactured from human plasma. Risk of transmitting infectious agents (e.g., HIV, hepatitis and, theoretically, Creutzfeldt-Jakob disease) has been greatly reduced by screening, testing, and manufacturing techniques. However, risk of transmission cannot be totally eliminated. ■ Hepatitis A and B vaccines are recommended for patients receiving plasma derivatives. ■ Thrombotic events have been reported. Use caution in patients with known risk factors for thrombosis. Incidence may be higher in females. ■ Inhibitors may develop with large or frequent doses; see Monitor.
Monitor:
Complex contains blood group isoagglutinins (anti-A and anti-B). When very large or frequently repeated doses are needed, as when inhibitors are present or when presurgical and postsurgical care is involved, patients of blood groups A, B, and AB should be monitored for signs of intravascular hemolysis and decreasing hematocrit values; see Antidote. ■ Replacement therapy should be monitored by appropriate coagulation tests, especially in cases involving major surgery. Monitor factor VIII and vWF:RCof as indicated in dosing guidelines.
Patient education:
Prophylactic hepatitis A and hepatitis B vaccines recommended. ■ Report symptoms of possibly transmitted viral infections immediately. Symptoms may include anorexia, arthralgias, fatigue, jaundice, low-grade fever, nausea, or vomiting. ■ Report rash or any other sign of hypersensitivity reaction promptly.
Maternal/child:
Category C: use only if clearly needed. ■ Adequate and well-controlled studies with long-term evaluation of joint damage have not been done in pediatric patients. Joint damage may result from suboptimal treatment of hemarthroses. ■ Safety and effectiveness for use in neonates with vWD has not been established. Has been used safely in infants, children, and adolescents with vWD.
Elderly:
Numbers insufficient to determine differences in response compared with younger adults. Consider overall status in dosing.
Drug/lab interactions
Specific information not available.
Side effects
Alphanate and Humate-P:
Usually well tolerated. Rare cases of hypersensitivity reactions, including anaphylaxis, have been reported (symptoms may include chest tightness, edema, fever, pruritus, rash, throat tightness). Other reported side effects include chills, headache, lethargy, nausea and vomiting, paresthesia, phlebitis, somnolence, and vasodilation. Inhibitors of factor VIII may occur.
Post-marketing:
Alphanate: In addition to the above, cardiac arrest, femoral venous thrombosis, flushing, itching, joint pain, pulmonary embolus, seizure, shortness of breath, swelling of the parotid gland, urticaria. Humate-P: Hypersensitivity reactions (including anaphylaxis), development of inhibitors to factor VIII, hemolysis, hypervolemia, thromboembolic complications.
Antidote
Keep physician informed of all side effects. If mild reactions occur (mild allergic reaction, chills, nausea, or stinging at the infusion site) and additional treatment is indicated, a product from a different lot should be considered. Discontinue immediately at first sign of a moderate to severe hypersensitivity reaction. Treat as necessary (antihistamines, epinephrine, corticosteroids). Development of acute hemolytic anemia, increased bleeding tendency, or hyperfibrinogenemia may require transfusion with Type O red blood cells. Discontinue administration of Alphanate/Humate-P and consider alternative therapy. Resuscitate as necessary.
Anti-inhibitor coagulant complex 
(an-TIE-in-HIH-bih-tor coe-AG-you-lant COM-plex)
Feiba
Antihemorrhagic
Usual dose
A unit of Feiba (nanofiltered and vapor heated) is expressed as factor VIII inhibitor bypassing activity. Identification of factor VIII inhibitor levels and PT mandatory before administration.
Feiba
Range is 50 to 100 units/kg.
Specific suggested dosing regimens include:
Joint hemorrhage:
50 units/kg; repeat at 12-hour intervals if indicated. May be increased to 100 units/kg if indicated. Continue treatment until clinical improvement (e.g., mobilization of the joint, reduction of swelling, relief of pain). Do not exceed 200 units/kg/24 hr.
Mucous membrane bleeding:
50 units/kg; repeat at 6-hour intervals if indicated. Monitor visible bleeding sites and hematocrit closely. May be increased to 100 units/kg every 6 hours for up to 2 doses if bleeding does not stop. Do not exceed 200 units/kg/24 hr.
Serious soft tissue hemorrhage:
100 units/kg; repeat at 12-hour intervals if indicated.
Other severe hemorrhage (e.g., CNS bleeding):
100 units/kg; repeat at 12-hour intervals if indicated. Feiba may be indicated at 6-hour intervals until clear clinical improvement occurs.
Dilution
Actual number of units shown on each vial. Use only the diluent provided and maintain strict aseptic technique. Bring to room temperature before reconstitution. Follow manufacturer’s guidelines for reconstitution using the BAXJECT device. May stick to sides of glass syringes; use of plastic syringes recommended. Gently swirl. Do not shake. May be given through Y-tube or three-way stopcock of infusion set. To avoid hypotension from prekallikrein activator (PKA), give Feiba within 3 hours of reconstitution.
Filters:
The manufacturer of Feiba uses a needleless transfer device and has no recommendation for use of an in-line filter.
Storage:
Within the indicated shelf life, may be stored in original packaging to protect from light at RT (not to exceed 25° C [77° F]). Do not freeze.
Compatibility
Specific information not available. Administration through a separate line without mixing with other IV fluids or medications is generally recommended. If anti-inhibitor coagulant is given as a continuous IV, one source says heparin 5 to 10 units/mL of concentrate may be added to avoid thrombophlebitis.
Rate of administration
If symptoms of too-rapid infusion (headache, flushing, changes in BP or pulse rate) occur, discontinue until symptoms subside. Restart at a lower rate.
Feiba:
Do not exceed 2 units/kg/min.
Actions
An activated prothrombin complex prepared from pooled human plasma. Controls bleeding in patients with factor VIII inhibitors. Mechanism of action is not well understood. Onset of response is usually within 12 hours. Peak response is usually seen within 36 to 72 hours.
Feiba:
1 unit of activity is the amount of anti-inhibitor coagulant complex (AICC) that will shorten the aPTT of a high-titer factor VIII inhibitor reference plasma to 50% of the blank value.
Indications and uses
Prophylaxis and treatment of hemorrhagic complications in hemophiliacs (hemophilia A or B) with factor VIII inhibitors who are bleeding or will undergo elective or emergency surgery. Anti-inhibitor coagulant complex (AICC) is most frequently indicated if presenting factor VIII inhibitor levels are above 5 to 10 Bethesda units (BU) or rise to that level following treatment with antihemophilic factor (AHF). Patients whose factor VIII inhibitor levels are between 5 and 10 BU and whose inhibitor levels remain at those levels may be treated with either AHF or AICC. Patients whose inhibitor levels are less than or equal to 5 BU and whose inhibitor levels remain at those levels may be treated with AHF.
Unlabeled uses:
Feiba has been used in the prophylaxis and treatment of hemorrhagic complications in nonhemophiliacs with acquired inhibitors to factors VIII, XI, and XII.
Contraindications
Patients with a normal coagulation mechanism; patients with significant signs of DIC; treatment of bleeding due to coagulation factor deficiencies in the absence of inhibitors to coagulation factors VIII or IX.
Precautions
Anamnestic responses (development of antibodies, reducing effectiveness of the drug) with a rise in factor VIII inhibitor titers have been seen in up to 20% of cases. ■ Thrombotic and thromboembolic events have been reported (e.g., DIC, venous thrombosis, pulmonary embolism, MI, and stroke). Risk of complications may be increased in surgical patients, in patients with thrombotic risk factors, and in patients receiving higher doses. Patients receiving more than 100 units/kg/dose or 200 mg/kg/day are at increased risk for DIC or acute coronary ischemia. Use high doses only as long as necessary to stop bleeding. ■ Patients with DIC, advanced atherosclerotic disease, crush injury, septicemia, or concomitant treatment with recombinant factor VIIa (e.g., NovoSeven RT) have an increased risk of developing thrombotic events due to circulating tissue factor (TF) or predisposing coagulopathy. ■ Use with caution and only if there are no therapeutic alternatives in patients at risk for DIC or arterial or venous thrombosis or in patients with existing thrombotic conditions (e.g., MI or venous thrombosis). ■ Use with caution in patients with a history of coronary heart disease, liver disease, or postoperative immobilization, in the elderly, and in neonates; weigh benefits versus risk. ■ Do not use Feiba for the treatment of bleeding episodes resulting from coagulation factor deficiencies. ■ Made from human plasma and may contain infectious agents (e.g., HIV, Creutzfeldt-Jakob disease, hepatitis B, or hepatitis C). Numerous steps in the manufacturing process are used to reduce the potential for infection. ■ See Drug/Lab Interactions.
Monitor:
Monitor PT before and after treatment. Use only accurate means of treatment evaluation; see Drug/Lab Interactions. ■ Monitor vital signs before, during, and after the infusion. ■ Monitor for S/S of acute coronary ischemia, DIC, and other thrombotic or thromboembolic events (e.g., changes in BP and HR, chest pain, cough, respiratory distress); see Antidote. ■ Laboratory indications of DIC may include decreased fibrinogen, decreased platelet count, and/or the presence of fibrin-fibrinogen degradation products (FDP) or significantly prolonged TT, PT, or PTT. ■ Monitor for S/S of a hypersensitivity reaction (e.g., rash, shortness of breath).
Patient education:
Manufactured from pooled human plasma. Possibility of viral transmission exists. Promptly report S/S of viral infections (e.g., chills, drowsiness, fever, runny nose followed by joint pain, rash, and/or S/S of hepatitis A [e.g., days to weeks of poor appetite, low-grade fever, and tiredness followed by abdominal pain, dark urine, jaundice, nausea, vomiting]).
Maternal/child:
Category C: use only if clearly needed. ■ Data not available for use in newborns. ■ Safety for use in breast-feeding not established.
Elderly:
May have increased risk for thromboembolic complications.
Drug/lab interactions
Not recommended for use with antifibrinolytic products (aminocaproic acid, tranexamic acid). Feiba has been used with antifibrinolytics; however, they should be used with caution and not administered until at least 12 hours after Feiba. ■ aPTT, WBCT, and other clotting factor tests do not correlate with clinical improvement. Attempts to normalize these values may lead to overdose and DIC.
Side effects
Anaphylaxis, bradycardia, chest pain, chills, cough, decreased fibrinogen concentration, decreased platelet count, DIC, fever, flushing, headache, hypertension, hypotension, myocardial infarction, prolonged PT, prolonged PTT, prolonged thrombin time, respiratory distress, tachycardia, thrombosis or thromboembolism, urticaria. Consider risk potential of contracting AIDS or hepatitis.
Overdose:
Increased risk for DIC, MI, or thromboembolism.
Post-marketing:
Anaphylactic reaction, DIC, hypersensitivity, hypoaesthesia, facial hypoaesthesia, hypotension, injection site pain, thromboembolism, thrombosis.
Antidote
If side effects occur, discontinue the infusion and notify the physician. May be resumed at a slower rate or discontinued, or an alternate product may be used. Symptoms of DIC (BP and pulse rate changes, respiratory discomfort, chest pain, cough, prolonged clotting tests, cyanosis of hands and feet, persistent bleeding from puncture sites or mucous membranes) require discontinuation of the infusion and immediate treatment. Treat anaphylaxis or other hypersensitivity reactions (changes in BP or HR [may indicate prekallikrein activity]) with antihistamines, epinephrine, corticosteroids and resuscitate as necessary.
Antithrombin III (human)
(an-tie-THROM-bin)
AT-III, Thrombate III
Anticoagulant
Antithrombotic
pH 6.5 to 7.5
Usual dose (international units [IU])
Loading dose, maintenance dose, and dosing intervals are completely individualized based on confirmed diagnosis (see Precautions), patient weight, clinical condition, degree of deficiency, type of surgery or procedure involved, physician judgment, desired level of antithrombin III (AT-III), and actual plasma levels achieved as verified by appropriate lab tests. One unit/kg should raise the level of AT-III by 1.4%. The desired AT-III level after the first dose should be about 120% of normal (normal is 0.1 to 0.2 Gm/L). AT-III levels must be maintained at normal or at least above 80% of normal for 2 to 8 days depending on individual patient factors. Usually achieved by administration of a maintenance dose once daily. Concomitant administration of heparin usually indicated; see Drug/Lab Interactions.
Calculate the initial loading dose using the following formula (assumes a plasma volume of 40 mL/kg):
Dosage units=(Desired AT-III level [%]-Baseline AT-III [%])×Body weight (kg)÷1.4%
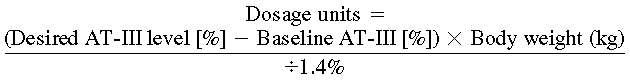
For a 70-kg patient with a baseline AT-III level of 57% the initial dose of Thrombate III would be (120% − 57%) × 70 ÷ 1.4 = 3,150 international units (IU). Measurement of plasma levels is suggested preinfusion, 20 minutes postinfusion (peak), 12 hours postinfusion, and preceding next infusion (trough). If recovery differs from the anticipated rise of 1.4% for each IU/kg, modify the formula accordingly. If the above patient has a 20-minute AT-III level of 147%, the increase in AT-III measured for each 1 IU/kg administered is (147% − 57%) × 70 kg ÷ 3,150 IU = 2% rise for each IU/kg administered. This in vivo recovery would be used to calculate future doses. A maintenance dose of approximately 60% of the loading dose every 24 hours is the average required to maintain plasma levels between 80% and 120%. Dose and interval based on plasma levels.
Dose adjustments
Dilution
Diluent, double-ended needles for dilution, and filter needle for aspiration into a syringe are provided. Warm unopened diluent and concentrate to room temperature. Enter diluent bottle with double-ended transfer needle first. Enter vacuum concentrate bottle with double-ended transfer needle at a 45-degree angle. Direct diluent from above to sides of vial to gently moisten all contents. Remove diluent bottle and transfer needle; swirl continuously until completely dissolved. Draw into a syringe through the filter needle. Remove filter needle; replace with an administration set (not provided). For larger doses, several bottles may be drawn into one syringe. Use a separate filter needle for each bottle.
Filters:
Filter needle supplied by manufacturer; see Dilution. For larger doses, several bottles may be drawn into one syringe. Use a separate filter needle for each bottle.
Storage:
Store in refrigerator before dilution; avoid freezing. Do not refrigerate after reconstitution. Use within 3 hours of reconstitution.
Compatibility
Administration through a separate line without mixing with other IV fluids or medications is recommended.
Rate of administration
Actions
Manufactured from human plasma, purified and heat treated through specific processes, AT-III is a plasma-based protein produced by the body to inactivate specific clotting proteins and control clot formation. Identical to heparin cofactor I, a factor in plasma necessary for heparin to exert its anticoagulant effect. It inactivates thrombin and the activated forms of factors IX, X, XI, and XII (all coagulation enzymes except factors VIIa and XIIIa). Increases AT-III levels within 30 minutes and has a half-life of up to 3 days.
Indications and uses
Treatment of patients with hereditary AT-III deficiency to prevent thrombosis during surgical or obstetric procedures (replacement therapy) or during acute thrombotic episodes.
Contraindications
None when used as indicated.
Precautions
For IV use only. ■ Confirm diagnosis of hereditary AT-III deficiency based on a clear family history of venous thrombosis as well as decreased plasma AT-III levels and the exclusion of acquired deficiency. Present laboratory tests may not be able to identify all cases of congenital AT-III deficiency. ■ Every unit of plasma used to manufacture AT-III is tested and found nonreactive for HBsAg and negative for antibody to HIV by FDA-approved tests, then heat-treated by a special process. Even with these precautions, individuals who receive multiple infusions may develop viral infection, particularly non-A, non-B hepatitis. HIV infection remains a remote possibility. ■ May reverse heparin resistance.
Monitor:
See varying methods for measuring AT-III levels under Usual Dose. Should be measured at least twice daily until the patient is stabilized and peak and trough levels established, then measured daily. All blood work should be drawn immediately before the next infusion of AT-III.
Patient education:
Inform of risks of thrombosis in connection with pregnancy and surgery and the fact that AT-III deficiency is hereditary.
Maternal/child:
Neonatal AT-III levels should be measured immediately after birth if parents are known to have AT-III deficiency (fatal neonatal thromboembolism [e.g., aortic thrombi] has occurred). Treatment of the neonate should be under the direction of a physician knowledgeable about coagulation disorders. Normal full-term and premature infants have lower than adult averages of AT-III plasma levels. ■ Category B: use only if clearly indicated. Fetal abnormalities not noted when administered in the third trimester. ■ Safety for use in pediatric patients not established.
Drug/lab interactions
Half-life of AT-III decreases with concurrent heparin treatment. The anticoagulant effect of heparin is enhanced, and a reduced dose of heparin and low-molecular-weight heparins (LMWHs) is indicated to avoid bleeding.
Side effects
Bowel fullness, chest pain, chest tightness, chills, cramps, dizziness, fever, film over eye, foul taste in mouth, hives, light-headedness, oozing and hematoma formation, and shortness of breath have occurred with Thrombate III. Some patients with acquired AT-III deficiency diagnosed with disseminated intravascular coagulation (DIC) have had diuretic and vasodilatory effects. Rapid infusion may cause dyspnea.
Antidote
Levels of 150% to 210% found in a few patients have not caused any apparent complications. Observe for bleeding. Reduce rate of infusion immediately for dyspnea. Decrease rate or interrupt infusion as indicated until side effects subside. Keep physician informed of patient’s lab values and condition.
Antithrombin recombinant
(an-tie-THROM-bin re-KOM-be-nant)
Atryn
Anticoagulant
Antithrombotic
pH 7
Usual dose (international units [IU])
Dose must be individualized for each patient and is based on the pretreatment level of functional antithrombin (AT) (expressed in percent of normal) and on body weight in kilograms according to the following chart. Treatment goal is to restore and maintain functional AT activity levels between 80% and 120% (0.8 to 1.2 IU/mL) of normal. Treatment should be initiated before delivery or approximately 24 hours before surgery to ensure AT level is in the target range. Different dosing formulas are used for the treatment of surgical and pregnant patients. Pregnant women being treated with antithrombin recombinant for any peripartum or perioperative event, including a cesarean section, should be treated according to the dosing formula for pregnant women.
| Antithrombin Recombinant Dosing Formula for Surgical Patients and Pregnant Women | |
| Loading Dose (IU) | Maintenance Dose (IU/hr) |
| Surgical Patients | |
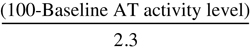 × Body weight (kg) × Body weight (kg) | 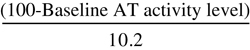 × Body weight (kg) × Body weight (kg) |
| Pregnant Women | |
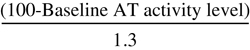 × Body weight (kg) × Body weight (kg) | 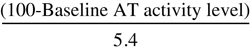 × Body weight (kg) × Body weight (kg) |

Check AT level just after surgery or delivery; AT activity may be rapidly decreased by surgery or delivery. If AT activity level is below 80%, administer an additional bolus dose to rapidly restore decreased AT activity level. Then restart the maintenance dose at the same rate of infusion as before the bolus. Monitor AT activity at least once or twice daily and adjust doses according to the chart in Dose Adjustments. Continue treatment until adequate follow-up anticoagulation is established.
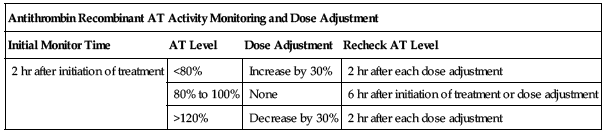
Dose adjustments
| Antithrombin Recombinant AT Activity Monitoring and Dose Adjustment | |||
| Initial Monitor Time | AT Level | Dose Adjustment | Recheck AT Level |
| 2 hr after initiation of treatment | <80% | Increase by 30% | 2 hr after each dose adjustment |
| 80% to 100% | None | 6 hr after initiation of treatment or dose adjustment | |
| >120% | Decrease by 30% | 2 hr after each dose adjustment | |
Dilution
Bring vials to RT no more than 3 hours before reconstitution. Each vial contains approximately 1,750 IU; exact potency is stated on the carton and label. Immediately before use, each vial must be reconstituted with 10 mL SWFI. Do not shake. Draw the reconstituted solution from one or more vials into a sterile syringe. May administer reconstituted solution directly or may further dilute in an infusion bag containing NS (e.g., dilute to obtain a final concentration of 100 IU/mL). Administer using an infusion set with a 0.22-micron in-line filter.
Filters:
Use of a 0.22-micron in-line filter required during infusion.
Storage:
Before use, refrigerate vials between 2° and 8° C (36° and 46° F). Use reconstituted or diluted solution within 8 to 12 hours of preparation. Do not use beyond expiration date on vial. Discard unused product.
Compatibility
Specific information not available. Because of specific use and unique formulation, consider administering through a separate line without mixing with other IV fluids or medications.
Rate of administration
Loading dose:
Administer as an infusion over 15 minutes. Follow immediately with the maintenance dose as a continuous infusion at the calculated IU/hr rate.
Actions
A recombinant human antithrombin produced by DNA technology. A DNA coding sequence for human antithrombin and a mammary gland–specific DNA sequence are introduced into genetically engineered goats. The goats’ milk contains the antithrombin. The amino acid sequence of antithrombin recombinant is identical to that of human plasma-derived antithrombin. Purified through numerous processes to eliminate potential viruses. AT is the principal inhibitor of thrombin and factor Xa. AT neutralizes the activity of thrombin and factor Xa by forming a complex that is rapidly removed from the circulation. When AT is bound to heparin, the ability of antithrombin to inhibit thrombin and factor Xa can be enhanced by greater than 300- to 1,000-fold. Half-life range based on IU/kg is 11.6 to 17.7 hours. This recombinant formulation has a shorter half-life and more rapid clearance compared with plasma-derived antithrombin (e.g., Thrombate III). Secreted in breast milk.
Indications and uses
Prevention of perioperative and peripartum thromboembolic events in patients with hereditary antithrombin deficiency. ■ Not indicated for treatment of thromboembolic events in patients with hereditary antithrombin deficiency.
Contraindications
Known hypersensitivity to goat and goat milk proteins.
Precautions
For IV use only. ■ Confirm diagnosis of hereditary antithrombin deficiency. ■ Hypersensitivity reactions may occur at any time during the infusion, thus requiring discontinuation of the infusion. ■ The anticoagulant effect of drugs that use antithrombin to exert their anticoagulation (e.g., heparin, low-molecular-weight heparins such as enoxaparin [Lovenox]) may be altered when antithrombin recombinant is added or withdrawn. Avoid excessive or insufficient anticoagulation by monitoring coagulation tests suitable for the anticoagulant used (e.g., aPTT and anti-factor Xa activity). To avoid bleeding or thrombosis, perform these tests regularly and at close intervals, especially during the first hours after the start or withdrawal of antithrombin recombinant. ■ See Drug/Lab Interactions.
Monitor:
Specific coagulation tests are required before administration and throughout the infusion process; see Usual Dose and Precautions. ■ Monitor throughout the infusion for S/S of a hypersensitivity reaction (e.g., hives, hypotension, generalized urticaria, tightness of the chest, wheezing, and/or anaphylaxis). ■ Monitor for S/S of bleeding or thrombosis.
Patient education:
Inform physician of a past or present allergy to goats or goat milk. ■ Promptly report S/S of a hypersensitivity reaction (e.g., rash, shortness of breath, wheezing). ■ Risk of bleeding increased when used with other anticoagulants. Report bleeding from any source.
Maternal/child:
Category C: use during pregnancy only if clearly needed. Studies have not shown that antithrombin recombinant increases the risk of fetal abnormalities if administered during the third trimester of pregnancy. Adverse reactions have not been reported in neonates born to women treated with antithrombin recombinant during clinical trials. ■ Indicated for prevention of thromboembolic events in women with hereditary antithrombin deficiency during labor and delivery. ■ Levels that appear in breast milk are estimated to be the same as in normal lactating women; however, use only if clearly needed and with caution during breast-feeding. ■ Safety and effectiveness for use in pediatric patients not established.
Elderly:
Numbers in clinical studies insufficient to determine if elderly patients respond differently than younger subjects. Dosing should be cautious in the elderly. Reduced doses may be indicated based on the potential for decreased organ function and concomitant disease or drug therapy.
Drug/lab interactions
The anticoagulant effect of heparin and low-molecular-weight heparin is enhanced by antithrombin. Concurrent use with these anticoagulants may alter the half-life of antithrombin. Concurrent use with heparin, low-molecular-weight heparins such as enoxaparin (Lovenox), or other anticoagulants that use antithrombin to exert their anticoagulant effect must be monitored clinically and biologically. To avoid excessive anticoagulation, perform regular coagulation tests (aPTT and, where appropriate, anti-factor Xa activity) at close intervals and adjust the dose of anticoagulant as indicated.
Side effects
Hemorrhage and infusion site reactions were most commonly reported. Hemorrhage may be serious (intra-abdominal, hemarthrosis, and postprocedural). Less common side effects include feeling hot, hematoma, hematuria, hepatic enzyme abnormalities, hypersensitivity reactions (including anaphylaxis), and noncardiac chest pain.
Antidote
Keep physician informed of patient’s lab values and condition. Discontinue the infusion if a hypersensitivity reaction occurs. ■ Treat anaphylaxis immediately with oxygen, epinephrine (Adrenalin), antihistamines (e.g., diphenhydramine [Benadryl]), vasopressors (e.g., dopamine), corticosteroids, albuterol, IV fluids, and ventilation equipment as indicated. Resuscitate as necessary.
Anti-thymocyte globulin (rabbit) 
(an-tie-THI-mo-cite GLOB-you-lin)
Thymoglobulin, Atgam (equine)*
Immunosuppressant
pH 7 to 7.4
Usual dose
Premedication:
To reduce the incidence and intensity of side effects during the infusion of anti-thymocyte globulin; premedication 1 hour before the infusion with corticosteroids (e.g., dexamethasone [Decadron]), acetaminophen (e.g., Tylenol), and/or an antihistamine (e.g., diphenhydramine [Benadryl]) is recommended.
Anti-thymocyte globulin:
1.5 mg/kg of body weight daily for 7 to 14 days. Given as an infusion into a high-flow vein. Used in conjunction with maintenance immunosuppression (e.g., tacrolimus [Prograf], mycophenolate [Cell-Cept]); see Drug/Lab Interactions.
Dose adjustments
Reduce dose by one-half if WBC count is between 2,000 and 3,000 cells/mm3 or if platelet count is between 50,000 and 75,000 cells/mm3. ■ Consider withholding dose or stopping anti-thymocyte therapy if WBC count falls below 2,000 cells/mm3 or platelets fall below 50,000 cells/mm3.
Dilution
Calculate the number of vials required (25 mg/vial); 5 mL of SWFI as diluent per vial is supplied. Drug and diluent must be warmed to room temperature before dilution. Absolute sterile technique required throughout dilution process. For each vial required use a new syringe and needle. Withdraw 5 mL of diluent and inject into lyophilized powder. Rotate vial gently until powder is completely dissolved. Do not shake. Each reconstituted vial contains 25 mg (5 mg/mL). Must be further diluted by transferring into 50 mL of infusion solution (saline or dextrose) for each 25 mg of anti-thymocyte globulin. Total volume is usually between 50 to 500 mL. Invert the infusion bag gently once or twice to mix the solution.
Filters:
Use of a 0.22-micron in-line filter recommended.
Storage:
Refrigerate and protect from light until removed to prepare for reconstitution. Do not freeze. Do not use after expiration date on vial. Use reconstituted vials within 4 hours. Use infusion solutions immediately. Discard unused drug.
Compatibility (underline indicates conflicting compatibility information)
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
Administration through a separate line without mixing with other IV fluids or medications is suggested because of specific use and potential for anaphylaxis.
One source suggests the following compatibilities:
Y-site:
Heparin, hydrocortisone sodium succinate (Solu-Cortef).
Rate of administration
Use of a high-flow vein and a 0.22-micron filter recommended. Well-tolerated and less likely to produce side effects (e.g., chills and fever) when administered at the recommended rate and the patient is premedicated.
Initial dose:
A total daily dose equally distributed over a minimum of 6 hours.
Subsequent doses:
A total daily dose equally distributed over a minimum of 4 hours.
Actions
A purified, pasteurized, gamma immune globulin, obtained by immunization of rabbits with human thymocytes. Mechanism of action not fully understood. May induce immunosuppression by T-cell depletion and immune modulation. Made up of a variety of antibodies that recognize key receptors on T-cells (those cells responsible for attacking and rejecting a foreign substance within the body). Anti-thymocyte globulin antibodies can inactivate and kill these T-cells, thus reversing the rejection process. May prevent organ loss and reduce the need for retransplantation. T-cell depletion is usually observed within a day of initiating thymoglobulin therapy. Half-life averages 2 to 3 days but the drug remains active, targeting the offending immune cells for days to weeks after treatment.
Indications and uses
Treatment of kidney transplant acute rejection in conjunction with concomitant immunosuppression.
Unlabeled uses:
Compassionate use in the treatment of acute rejection in bone marrow, heart, and liver transplants. Treatment of myelodysplastic syndrome (MDS).
Contraindications
Patients with a known allergy to rabbit proteins, an acute viral illness, or a history of anaphylaxis during rabbit immunoglobulin administration.
Precautions
Administered only under the direction of a physician experienced in immunosuppressive therapy and management of renal transplant patients in a facility with adequate laboratory and supportive medical resources. ■ Not considered effective for treating antibody-mediated (humoral) rejections. ■ Prolonged use or overdose in combination with other immunosuppressive agents may cause over-immunosuppression resulting in severe infections and may increase the incidence of lymphoma or posttransplant lymphoproliferative disease (PTLD) or other malignancies. Use of appropriate antiviral, antibacterial, antiprotozoal, and/or antifungal prophylaxis is recommended. In clinical trials, viral prophylaxis with ganciclovir infusion was used. ■ In clinical trials, anti-rabbit antibodies developed in 68% of patients. Controlled studies on repeat use of anti-thymocyte globulin in patients with anti-rabbit antibodies have not been conducted. Use caution if repeat courses are indicated; monitoring of lymphocyte count is recommended to ensure that T-cell depletion is achieved. ■ If anaphylaxis occurs during or after therapy, further administration of anti-thymocyte globulin is contraindicated.
Monitor:
Close clinical observation is imperative. Monitor for side effects during and after infusion. Anaphylaxis has occurred; emergency equipment, medications, and supplies must be available. ■ Obtain baseline and monitor WBC and platelet counts during therapy. Thrombocytopenia or neutropenia may occur and are reversible following dose adjustment; see Dose Adjustments. ■ Monitoring of the lymphocyte count (i.e., total lymphocyte count and T-cell counts [absolute and/or subset]) may help assess the degree of T-cell depletion. ■ Monitor carefully for signs of infection. ■ Prophylactic antibiotics may be indicated pending results of C/S in a febrile neutropenic patient. ■ See Precautions and Drug/Lab Interactions.
Patient education:
Imperative that all medications (especially immunosuppressants) be reviewed with physician. ■ Report any previous hypersensitivity/anaphylactic reaction. ■ Report acute viral infections immediately. ■ Promptly report chest pain, irregular or rapid heartbeat, shortness of breath, swelling of the face or throat, or wheezing during infusion of medication. ■ See Appendix D, p. 1333. ■ May be associated with an increased risk of malignancy.
Maternal/child:
Category C: safety for use during pregnancy and breast-feeding not established. Safety and effectiveness for use in pediatric patients not established. Use only if clearly needed. ■ Has been used in pediatric patients in limited European studies and in the United States for compassionate use. Response similar to adults.
Elderly:
Specific information not available.
Drug/lab interactions
Concurrent use with immunosuppressants (e.g., azathioprine, cyclosporine [Sandimmune], mycophenolate [Cell-Cept], tacrolimus [Prograf]) may potentiate the immunosuppressive action of these agents; many transplant centers decrease maintenance immunosuppression therapy during the period of antibody therapy. ■ May stimulate the production of antibodies, which cross-react with rabbit immune globulins. ■ May interfere with rabbit antibody-base immunoassays and with cross-match or panel-reactive antibody cytotoxicity assays.
Side effects
Are dose-limiting. Abdominal pain, asthenia, diarrhea, dizziness, dyspnea, fever, headache, hyperkalemia, hypertension, infection, infusion reaction (e.g., chills and fever), leukopenia, malaise, nausea, pain, peripheral edema, tachycardia, and thrombocytopenia were reported frequently. Anaphylaxis has been reported.
Overdose:
Leukopenia or thrombocytopenia.
Antidote
Notify physician of all side effects. Most can be managed symptomatically. Manage leukopenia or thrombocytopenia during therapy or in overdose with dose reduction. Infusion reactions are managed with premedication and reduction in the rate of infusion. Treat infections aggressively; see Precautions. May require discontinuation of therapy. Discontinue infusion and/or therapy immediately if anaphylaxis occurs. Treat anaphylaxis immediately with epinephrine (Adrenalin), diphenhydramine (Benadryl), oxygen, vasopressors (e.g., dopamine), corticosteroids, IV fluids, and ventilation equipment as indicated. Resuscitate as necessary.
Antivenin crotalidae polyvalent immune fab (ovine)
(an-tee-VEN-in kro-TAL-ih-day pol-ih-VAY-lent im-MYOUN fab)
CroFab
Antivenin
Usual dose
Contact a regional poison control center for individual treatment advice.
Premedication:
Premedication may be indicated for patients with allergies. Obtain blood work before administration; see Contraindications, Precautions, and Monitor. Initiate as soon as possible after crotalid snakebite in patients who develop signs of progressive envenomation (e.g., worsening local injury, coagulation abnormality, or systemic signs of envenomation); see Monitor. Has been effective when given within 6 hours of snakebite.
Initial dose:
Skin testing for sensitivity to serum is not required. 4 to 6 vials is the recommended initial dose based on clinical experience. Adjust based on severity of envenomation when patient is initially assessed. Observe for up to 1 hour after initial dose administered. Desired outcome is complete control of the envenomation (i.e., complete arrest of local manifestations and return of coagulation tests and systemic signs to normal).
Repeat doses:
If control of symptoms is not accomplished by the initial dose, give an additional dose of 4 to 6 vials. This dose may be repeated until initial control of the envenomation syndrome has been achieved. After initial control has been established, give additional 2-vial doses every 6 hours for up to 18 hours (3 doses). Optimal dosing following the 18-hour scheduled dose has not been determined. Additional 2-vial doses may be given as directed by the treating physician, based on the patient’s clinical response. Up to 18 vials have been given without any observed direct toxic effect. Scheduled dosing rather than PRN dosing may provide better control of envenomation symptoms caused by the continued leaking of venom from depot sites.
Pediatric dose
Absolute venom dose following snakebite is expected to be the same in pediatric patients and adults; no dose adjustment for age is required. See Maternal/Child.
Dose adjustments
Dilution
Reconstitute each vial with 18 mL NS. Mix by continuous gentle swirling. Do not shake. Further dilute the contents of the reconstituted vials in 250 mL NS and mix by gently swirling.
Storage:
Refrigerate unopened vials; do not freeze. Reconstituted vials and diluted solution must be used within 4 hours.
Compatibility
Specific information not available. Administration through a separate line without mixing with other IV fluids or medications is suggested because of specific use and potential for anaphylaxis.
Rate of administration
Initiate infusion at a rate of 25 to 50 mL/hr for the first 10 minutes. Carefully observe for hypersensitivity reactions. If no adverse reaction occurs, increase rate to 250 mL/hr so that total dose is administered over 1 hour. Reduce rate of administration if infusion-related reactions occur (e.g., fever, low back pain, nausea, wheezing), and monitor closely.
Actions
A venom-specific Fab fragment of immunoglobulin G (IgG) obtained from the blood of healthy sheep flocks immunized with one of four snake venoms; see Indications for specific venoms. To obtain the final product, the four different monospecific antivenins are mixed. Works by binding and neutralizing venom toxins, facilitating their redistribution away from target tissues and their elimination from the body. Half-life is estimated to be 12 to 23 hours.
Indications and uses
Management of patients with North American Crotalid envenomation. The term Crotalid is used to describe the Crotalinae subfamily (formerly known as Crotalidae) of venomous snakes and includes rattlesnakes, copperheads, cottonmouths/water moccasins. Early use (within 6 hours) is advised to prevent clinical deterioration and the occurrence of systemic coagulation abnormalities.
Contraindications
Known history of hypersensitivity to sheep, papaya, or papain unless benefits outweigh risks and appropriate management for anaphylactic reactions is readily available.
Precautions
Usually administered in the hospital by or under the direction of the physician specialist with adequate diagnostic and treatment facilities readily available. ■ Patients with allergies to dust mites, latex, papain, chymopapain, other papaya extracts, the pineapple enzyme bromelain, or sheep protein may be at risk for a hypersensitivity reaction to this antivenin; see Contraindications; use caution. ■ Contains 0.03 mg of mercury per vial (ethyl mercury from thimerosal). Exposure from 18-vial dose is 0.6 mg of mercury. Definitive data not available; literature suggests that information related to methyl mercury toxicities may be applicable. ■ Recurrent coagulation abnormalities (e.g., decreased fibrinogen, decreased platelets, and elevated PT), defined as the return of a coagulation abnormality after it has been successfully treated with antivenin, were observed in patients who experienced coagulation abnormalities during their initial hospitalization and occurred in approximately one half of patients studied. Crotalidae immune fab has a shorter persistence in the blood than crotalid venoms, which can leak from depot sites over a prolonged period of time; repeat dosing to prevent or treat such recurrence may be necessary. Optimal dosing to prevent recurrent coagulopathy has not been determined. ■ Use caution if a repeat course of treatment is required for a subsequent envenomation episode; crotalidae immune fab is a foreign protein and antibodies may develop, producing sensitivity.
Monitor:
Before antivenin is administered, draw adequate blood for baseline studies (e.g., type and cross-match, CBC, hematocrit, platelet count, PT, bleeding and coagulation times, BUN, electrolytes, and bilirubin). ■ Severity of envenomation is based on six body categories: cardiovascular, gastrointestinal, hematologic, local wound (e.g., pain, swelling, and ecchymosis), nervous system, and pulmonary effects. Specific parameters of minimal, moderate, and severe envenomation are outlined in the following chart.
| Definition of Minimal, Moderate, and Severe Envenomation Used in Clinical Studies of Crotalidae Polyvalent Immune Fab | |
| Envenomation Category | Definition |
| Minimal | Swelling, pain, and ecchymosis limited to the immediate bite site. Systemic signs and symptoms absent. Coagulation parameters normal with no clinical evidence of bleeding. |
| Moderate | Swelling, pain, and ecchymosis involving less than a full extremity or, if bite was sustained on the trunk, head, or neck, extending less than 50 cm. Systemic signs and symptoms may be present but not life-threatening, including but not limited to nausea, vomiting, oral paresthesia or unusual tastes, mild hypotension (systolic BP less than 90 mm Hg), mild tachycardia (HR less than 150), and tachypnea. Coagulation parameters may be abnormal, but no clinical evidence of bleeding present. Minor hematuria, gum bleeding, and nosebleeds are allowed if they are not considered severe in the investigator’s judgment. |
| Severe | Swelling, pain, and ecchymosis involving more than an entire extremity or threatening the airway. Systemic signs and symptoms are markedly abnormal, including severe alteration of mental status, severe hypotension, severe tachycardia, tachypnea, or respiratory insufficiency. Coagulation parameters are abnormal, with serious bleeding or severe threat of bleeding. |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


