CHAPTER 8 Endocrinologic disorders
Endocrine assessment
1. Critical illness initiates the stress response.
2. The stress response increases the metabolic rate.
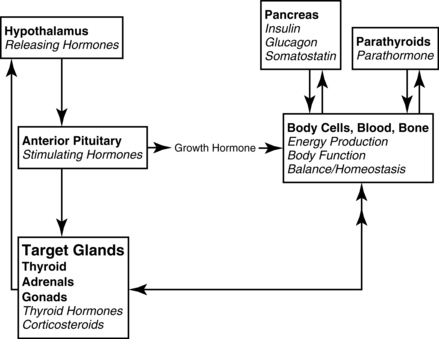
3. The hypothalamic-pituitary axis (Figure 8-1) regulates the metabolic rate. The thyroid and adrenal glands are extremely stressed by the stimulus of critical illness to maintain the increased metabolic rate, along with meeting the energy demands at the cellular level. Hypofunction of the thyroid and adrenals requires assessment of not only the primary glands, but also the hypothalamus (produces releasing factors) and anterior pituitary (produces stimulating hormones.) The hypothalamic-pituitary-target organ feedback loop must be fully intact to maintain normal metabolism.
4. The stress response markedly increases endogenous glucocorticoids, which increase blood glucose. The pancreas may not be able to produce sufficient insulin to manage the glucose level, resulting in hyperglycemia. Insulin may be required to manage hyperglycemia until the stress of illness resolves. People with diabetes mellitus are always challenged with hyperglycemia, and with additional illness, can experience the crisis states of diabetic ketoacidosis (DKA) and hyperosmolar hyperglycemic syndrome (HHS).
5. Under prolonged extreme stress, both the adrenal glands and thyroid may be unable to sustain hormone production to support the stress level. Supplemental glucocorticoids (corticosteroids) and thyroid hormones may be needed.
6. The assessment of the critically ill patient should focus on the signs of failure of the endocrine system to support the stress response. Signs of hypofunction are explained the sections on hyperglycemia, adrenal crisis, and myxedema coma. Patients with adequate function of the pancreas, thyroid, and adrenal glands under normal conditions may not be able to maintain balance when exposed to the stress of critical illness. Those with underlying hypofunction are more likely to experience crises.
7. Hyperthermia and cardiac symptoms can make diagnosis of thyroid storm difficult, as the crisis mimics other cardiac crises and infection. Thyroid storm results from underlying Graves’ disease/hyperthyroidism; not a complication of critical illness.
8. ![]() Assessment of the causes of unusual fluid and electrolyte imbalances should include evaluation of posterior pituitary function, in addition to screening for renal dysfunction. Posterior pituitary dysfunction may result in abnormal levels of antidiuretic hormone (ADH) and may be a complication of critical illness, or result from hypothalamic or pituitary disease. Diabetes insipidus (DI ) produces dehydration and Syndrome of Inappropriate ADH (SIADH) produces hyponatremia, which can reach critical states if not properly addressed. Nephrogenic DI results from failure of the kidneys to respond to ADH. The elderly are at higher risk of complications with DI as a result of age-related changes to the thirst mechanism and renal function.
Assessment of the causes of unusual fluid and electrolyte imbalances should include evaluation of posterior pituitary function, in addition to screening for renal dysfunction. Posterior pituitary dysfunction may result in abnormal levels of antidiuretic hormone (ADH) and may be a complication of critical illness, or result from hypothalamic or pituitary disease. Diabetes insipidus (DI ) produces dehydration and Syndrome of Inappropriate ADH (SIADH) produces hyponatremia, which can reach critical states if not properly addressed. Nephrogenic DI results from failure of the kidneys to respond to ADH. The elderly are at higher risk of complications with DI as a result of age-related changes to the thirst mechanism and renal function.
9. ![]() Medication noncompliance for management of existing endocrine disease must be assessed. People with lower income are at higher risk of crisis due to inability to purchase medications and teenagers may not practice meticulous management; particularly those with diabetes mellitus. Elders may not take medications appropriately due to lack of understanding or mis-dosing due to deteriorating short-term memory.
Medication noncompliance for management of existing endocrine disease must be assessed. People with lower income are at higher risk of crisis due to inability to purchase medications and teenagers may not practice meticulous management; particularly those with diabetes mellitus. Elders may not take medications appropriately due to lack of understanding or mis-dosing due to deteriorating short-term memory.
Acute adrenal insufficiency (adrenal crisis)
Pathophysiology
1. Stimulate the pituitary to secrete stimulating hormones (adrenocorticotropic hormone [corticotropin] [ACTH]), which in turn
2. Stimulates the adrenal glands to secrete glucocorticoids (cortisol) and mineralocorticoids (aldosterone), which in turn
3. Facilitates cellular functions throughout the body (metabolism), which in turn
4. Creates signals back to the hypothalamus from the cells to increase or decrease releasing hormones (see Figure 8-1).
If primary adrenal insufficiency is the cause of the crisis, the adrenal glands are the root cause of the problem. Addison disease manifests when the entire adrenal cortex is destroyed, which stops the production of glucocorticoids (cortisol) and mineralocorticoids (aldosterone). Glucocorticoids are essential hormones produced by the adrenal cortex that help maintain vascular tone and cardiac contractility, facilitate wound healing, and support immunity. Cortisol deficiency intensifies the clinical effects of hypovolemia by promoting a decrease in vascular tone, which is partially related to unopposed endothelial production of nitric oxide, and a decreased vascular response to the catecholamine hormones epinephrine and norepinephrine. Relative hypoglycemia may be present, as the breakdown of stored glycogen is not possible without cortisol. Mineralocorticoid hormones are primary regulators of fluid and electrolyte balance, and when unavailable, patients experience hyponatremia, hypovolemia, hyperkalemia, and metabolic acidosis. Large amounts of sodium and water are excreted in the urine. Severe hypotension, shock, and eventually death may occur without intravenous adrenocortical hormone and fluid replacement. In patients with chronic primary adrenocortical insufficiency or Addison disease, acute crises may be prevented by tripling hormone replacement doses during periods of stress.
8-1 RESEARCH BRIEF
The results were dismal in terms of mortality reduction because there was little difference from placebo. Of note, however, was that the group receiving hydrocortisone spent less time in shock by the prior mentioned definition.
From Sprung CL, Annane D, Keh D, et al: Hydrocortisone therapy for patients in septic shock. N Engl J Med 358(2):111–124, 2008.
Endocrine assessment adrenals
Goal of system assessment:
To evaluate for severe hypotension, refractory to volume and vasopressor administration
History and risk factors
• Extreme emotional or physiologic stress, which increases the need for adrenocortical “stress” hormones to mediate the stress response
• Patients previously receiving glucocorticoids (steroids) who may be abruptly withdrawn from steroids or are not given sufficient steroids to manage additional stress
• Adrenalectomy, hypophysectomy, sepsis, human immunodeficiency (HIV) disease
• Other medications: beta adrenergic blockers, diuretics, angiotensin-converting enzyme (ACE) inhibitors, angiotensin release blockers (ARBs), nitrates, aspirin, and other platelet inhibitors
Observation and vital signs: primary (first-degree) and secondary (second-degree) insufficiency
• Refractory, severe hypotension resulting from vascular collapse
• Acute abdomen assessment findings: abdominal pain, distention
• Hyperpyrexia, with temperatures often reaching in excess of 105°F
• Cyanosis, confusion; may be comatose
• Relative hypoglycemia: tremors, diaphoresis, tachycardia, tachypnea
Observation and vital signs: primary (first-degree) insufficiency only
• Prominent nausea and vomiting
• Signs of dehydration: poor skin turgor, sunken, soft eyeballs, weight loss
• Bronze hue to the skin secondary to excess production of ACTH
• Hyperkalemia, which may be associated with metabolic acidosis: peaked T waves, widening QRS complex, prolonged PR interval, flattened-to-absent P wave
Screening labwork
For suspected acute adrenal crisis
• Random plasma cortisol level: Drawn prior to initiating hydrocortisone replacement but must be interpreted with caution in critically ill patients, because greater than 90% of circulating cortisol is protein bound. When patients are hypoproteinemic, with serum albumin less than 2.5 g/dl, low values may be gleaned from all cortisol testing in patients who have normal adrenal function. A random plasma cortisol level of greater than 25 mcg/dl excludes both primary and secondary adrenal insufficiency.
• Free cortisol level: Done in the setting of hypoproteinemia to better determine the cortisol level. An abnormal test may require a complete endocrinology assessment when the patient stabilizes.
For noncritical adrenocortical insufficiency
• Corticotropin (ACTH) stimulation test: The goal is to differentiate primary from secondary adrenocortical insufficiency or to assess if the adrenal cortex is capable of producing cortisol. Testing of the hypothalamic-pituitary-adrenal axis using this test can differentiate primary from secondary insufficiency. Baseline plasma cortisol level is drawn immediately prior to ACTH administration.
• Adrenal insufficiency is diagnosed when:
Diagnostic Tests for Acute Adrenal Insufficiency
| Test | Purpose | Abnormal Findings |
|---|---|---|
| Noninvasive | ||
| Chest radiograph | Assess for heart size and presence of opportunistic infections (primary) | The chest radiogram may be normal but often reveals a small heart. Stigmata of earlier infection or current evidence of tuberculosis (TB) or fungal infection may be present when this is the cause of Addison disease. |
| Computed tomography (CT) scan of abdomen | Assess abdominal organs, size, presence of blood (primary) | Abdominal CT scan may be normal but may show bilateral enlargement of the adrenal glands in patients with Addison disease because of TB, fungal infections, adrenal hemorrhage, or infiltrating diseases involving the adrenal glands. In Addison disease due to TB or histoplasmosis, evidence of calcification involving both adrenal glands may be present. In idiopathic autoimmune Addison disease, the adrenal glands usually are atrophic. |
| Blood Studies | ||
| Complete blood count (CBC) Hemoglobin (Hgb) Hematocrit (Hct) RBC count (RBCs) WBC count (WBCs) | Assess for anemia, inflammation and infection (primary and secondary) | CBC count may reveal a normocytic normochromic anemia, which, upon initial presentation, may be masked by dehydration and hemoconcentration. Relative lymphocytosis and eosinophilia may be present. |
| Electrolytes Potassium (K+) Sodium (Na+) | Assess for abnormalities of aldosterone (primary) | Elevation in K+ may cause dysrhythmias; decrease of Na+ may indicate fluid retention and/or concomitant heart failure. |
| Serum glucose | Assess for relative hypoglycemia (primary and secondary) | Hypoglycemia may be present in fasted patients, or it may occur spontaneously. It is caused by the increased peripheral utilization of glucose and increased insulin sensitivity. It is more prominent in children and in patients with secondary adrenocortical insufficiency. |
| Thyroid-stimulating hormone | Assess for thyroid dysfunction (primary and secondary) | Increased thyroid-stimulating hormone, with or without low thyroxine, with or without associated thyroid autoantibodies, and with or without symptoms of hypothyroidism, may occur in patients with Addison disease and in patients with secondary adrenocortical insufficiency due to isolated ACTH deficiency. These findings may be reversible with cortisol replacement. |
Collaborative management
DIAGNOSIS AND MANAGEMENT OF CORTICOSTEROID INSUFFICIENCY IN CRITICALLY ILL PATIENTS
| Recommendations for the diagnosis and management of corticosteroid insufficiency in critically ill adult patients: consensus statements from an international task force by the American College of Critical Care Medicine. | |
| In 2008, an interdisciplinary, multispecialty task force of experts in critical care medicine was convened from the membership of the Society of Critical Care Medicine and the European Society of Intensive Care Medicine. In addition, international experts in endocrinology were invited to participate. The goal was to develop a strategic tool for defining and treating critical illness acute adrenal insufficiency. | |
| Treatment | Rationale |
| Moderate dose of hydrocortisone (200–300 mg/day) for critically ill patients with septic shock. | Six randomized control trials demonstrate significant and greater shock reversal in patients who received hydrocortisone although no difference in mortality. |
| Moderate dose of hydrocortisone in the management of severe early ARDS (PF ratio <200) instituted before day 14. | Five randomized studies evaluated moderate hydrocortisone administration in ARDS from various origins. Consistent improvement was reported in the PF ratio, inflammatory markers were reduced, and both ventilator days and ICU length of stay were reduced. |
| In patients with septic shock, intravenous hydrocortisone should be given in a dose of 200 mg/day in four divided doses or as a bolus of 100 mg followed by a continuous infusion at 10 mg/hr (240 mg/day).The optimal initial dosing regimen in patients with early severe ARDS is 1 mg/kg/day methylprednisolone as a continuous infusion. | Multiple clinical trials, both randomized and not, as well as prospective and retrospective of patients in severe sepsis and ARDS. |
| Glucocorticoid (GC) treatment should be tapered slowly and not stopped abruptly. | Abruptly stopping hydrocortisone will likely result in a rebound of proinflammatory mediators, with recurrence of the features of shock (and tissue injury). |
| Treatment with dexamethasone has previously been suggested in patients with septic shock until an ACTH stimulation test is performed, this approach can no longer be endorsed | Physiologic and pathologic understanding that dexamethasone leads to immediate and prolonged suppression of ACTH. |
From Marik PE, Pastores SM, Annane D, et al: Crit Care Med 36(6):1937–1949, 2008.
Care priorities
Use vasopressors if intravascular volume replacement fails to effectively increase BP (see Appendix 6). Response to catecholamine infusions (epinephrine, norepinephrine, dopamine) is reduced in adrenal insufficiency; higher-than-normal doses may be needed to manage refractory hypotension.
• Administer 100 mg of hydrocortisone in 100 ml of normal saline solution by continuous IV infusion at a rate of 12 ml/hr. Infusion may be initiated with 100 mg of hydrocortisone as an IV bolus.
• A continuous infusion method maintains plasma cortisol levels effectively if the stress level is steady or constant, particularly in patients who rapidly metabolize the drug. Rapid metabolizers have a greater likelihood of having low plasma cortisol levels between IV boluses.
• An alternative method of hydrocortisone administration is 50-75 mg IV every 4-6 hours for 5 days.
• Improvement in BP and other vital signs should be evident within 4 to 6 hours of hydrocortisone infusion. If not, the diagnosis of adrenal insufficiency is questionable.
• After 2 to 3 days, the stress hydrocortisone dose should be reduced to 100 to 150 mg, infused over a 24-hour period regardless of the patient’s status. In addition to helping with adrenal recovery, lower doses may help abate gastrointestinal (GI) bleeding.
• As the patient improves and the clinical situation allows, the hydrocortisone infusion can be gradually tapered over the following 4 to 5 days to daily replacement doses of approximately 3 mg/hr (72 to 75 mg over 24 hours) and eventually to daily oral replacement doses when oral intake is possible.
• If the patient receives at least 100 mg of hydrocortisone in 24 hours, no mineralocorticoid replacement is necessary, because the mineralocorticoid activity of hydrocortisone in this dosage is sufficient.
• As the hydrocortisone dose continues to be weaned, mineralocorticoid replacement should begin in doses equivalent to the daily adrenal gland aldosterone output of 0.05 to 0.1 mg daily or every other day.
4. Maintain normal blood glucose level:
If patient is initially hypoglycemic, 50% dextrose may be needed to correct hypoglycemia. When hydrocortisone or other cortisol replacement is initiated, hyperglycemia may result. An insulin infusion may be needed to control the blood glucose (see Hyperglycemia,p 711).
CARE PLANS FOR ADRENAL INSUFFICIENCY
![]()
Fluid and electrolyte management
1. Monitor vital signs and hemodynamic measurements every 15 minutes until stabilized for 1 hour. Consult physician or midlevel practitioner promptly for deterioration in vital signs or hemodynamics.
2. Administer IV fluids to replace fluid volume. Initially, rapid fluid replacement is essential.
3. Maintain accurate input and output (I&O) record. Weigh patient daily.
4. Monitor for electrolyte imbalance. Imbalances associated with adrenal insufficiency include the following:
5. Monitor ECG continuously; observe for potassium-related changes. Increased ventricular irritability may signal hypokalemia. (See Fluid and Electrolyte Disturbances, Hypokalemia, p 52.)
6. Monitor laboratory results. With appropriate treatment, serum sodium levels should rise to normal and serum potassium levels should fall to normal. Prevent rapid correction or overcorrection of hyponatremia. Serum sodium levels should not be allowed to increase greater than 12 mEq/L during the first 24 hours of treatment because of the risk of neurologic damage. (See Fluid and Electrolyte Disturbances, Hyponatremia, p 46, or Syndrome of Inappropriate ADH, p 734.)
7. Assess mental and respiratory status at frequent intervals. Institute safety measures as indicated. Reorient and reassure patient as needed.
8. Encourage oral fluid intake as patient’s condition stabilizes. Add sodium-rich foods (see Box 8-1) as tolerated. Begin oral glucocorticoid replacement therapy as prescribed.
9. Consult physician or midlevel practitioner if signs and symptoms of fluid and/or electrolyte imbalance persist or worsen.
Box 8-1 PATIENT AND FAMILY EDUCATION CONCERNING GLUCOCORTICOID AND MINERALOCORTICOID REPLACEMENT
Glucocorticoids (e.g., cortisone, acetate, prednisone)
• Take medication in a diurnal pattern to mimic normal secretion (i.e., two thirds of dose in the morning and one third of dose in the afternoon).
• Take steroids with food to decrease gastric irritation.
• Weigh self regularly, and report to physician gains of greater than 2 lb/wk.
• Avoid exposure to infection, and be alert to indicators of infection (e.g., fever, nausea, diarrhea, malaise).
• Contact physician promptly during periods of physical or emotional stress; dosages will require adjustment at these times.
• Indicators of overreplacement: weight gain (moon face, truncal obesity); edema, thin, fragile skin (striae, easy bruising); slow wound healing; chronic fatigue; emotional lability
• Indicators of underreplacement: weight loss, hyperpigmentation, skin creases, anorexia, nausea, abdominal discomfort, chronic fatigue, depression, irritability
Mineralocorticoids (e.g., fludrocortisone, desoxycorticosterone acetate)
• As prescribed, modify diet with liberal amounts of sodium (see Box 1-4), protein, and carbohydrates.
• Weigh self regularly, and report to physician sudden gains or losses greater than 2 lb/wk.
• Contact physician promptly during periods of physical or emotional stress; dosages will require adjustment at these times.
• Indicators of overreplacement: edema, muscle weakness, hypertension
• Indicators of underreplacement: excessive urination, weight loss, decreased skin turgor
![]()
Immune Status; Infection Status; Energy Conservation
1. Monitor and report signs of increasing crisis: urinary output increased from usual amount, changes in LOC, orthostatic hypotension, nausea, vomiting, and tachycardia.
2. Provide a quiet, nonstressful environment. Adjust lighting to meet needs of individual activities, avoiding direct light in the eyes. Control noise when possible. Prevent unnecessary interruptions, and allow for rest periods. Limit the number of visitors and the length of time they spend with patient. Speak softly and reassuringly to patient.
3. Monitor for and manage hyperthermia using tepid baths, antipyretics, and cooling blankets.
4. Maintain a cool environmental temperature. Maintain strict environmental asepsis, and monitor patient carefully for signs of infection. Avoid exposing patient to staff members or visitors who have colds or infections.
![]() Fluid Monitoring; Environmental Management; Infection Protection
Fluid Monitoring; Environmental Management; Infection Protection
Deficient knowledge: illness care
![]()
Knowledge: Disease Process; Knowledge: Energy Conservation; Knowledge: Medication
1. Teach patient about prescribed medications, including purpose, dosage, route of administration, and potential side effects (Box 8-1). Medication administration should mimic normal diurnal pattern of plasma cortisol levels (e.g., two thirds in the morning and one third in late afternoon).
2. Provide dietary instruction: dietary sodium and potassium may need to be adjusted on the basis of the patient’s clinical condition and drug therapy (see discussions of sodium and potassium in Fluid and Electrolyte Disturbances, p. 37).
3. Explain the importance of controlling stress, both emotional and physiologic, which increases adrenal demand. Teach patient to seek medical intervention during times of increased stress (e.g., fever, infection), inasmuch as medication dosages may need to be increased.
4. Teach indicators of overreplacement and underreplacement of steroids, which require prompt medical attention (see Box 8-1).
5. Stress the importance of never abruptly discontinuing use of any steroid preparation. Use must be tapered to avoid precipitation of crisis.
6. Remind patient of the importance of continued medical follow-up.
7. Explain the procedure for obtaining a medical-alert bracelet or card.
Diabetes insipidus
Pathophysiology
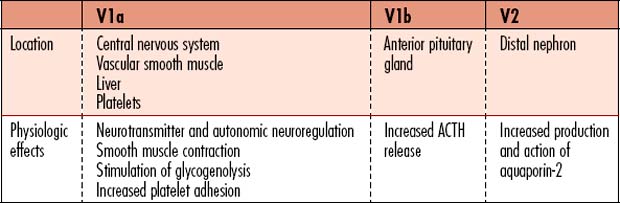
Diabetes insipidus (DI) is a metabolic disorder that affects total body free water regulation, resulting in an abnormally high output of extremely dilute urine, increased fluid intake, and constant thirst. The volume of hypotonic urine excreted is 3-20L/day. Vasopressin (antidiuretic hormone [ADH]) is a key component in the regulation of fluid and electrolyte balance, through direct effects on renal water regulation. Vasopressin is produced in the hypothalamus, is stored in the posterior pituitary gland, and exerts action in the kidneys for water regulation. Three subtypes of receptors respond to the effects of vasopressin (Table 8-1).
• Rapid changes of plasma osmolality: Rapid increases in plasma osmolality result in an abnormal increase in ADH/vasopressin release.
• Drinking fluids: Oral fluid consumption rapidly suppresses the release of ADH, through afferent pathways originating in the oropharynx.
• Pregnancy: The osmotic threshold for ADH release is lowered in pregnancy.
• Aging: ![]() Plasma vasopressin concentrations increase with age, together with enhanced ADH responses to osmotic stimulation. Age-related changes in ADH production can result in blunting of the thirst response, decreased fluid intake, impaired ability to excrete a free water load, and reduced ability of the kidneys to concentrate urine. These changes predispose the elderly to both hypernatremia and hyponatremia.
Plasma vasopressin concentrations increase with age, together with enhanced ADH responses to osmotic stimulation. Age-related changes in ADH production can result in blunting of the thirst response, decreased fluid intake, impaired ability to excrete a free water load, and reduced ability of the kidneys to concentrate urine. These changes predispose the elderly to both hypernatremia and hyponatremia.
• Central, hypothalamic or pituitary DI (neurogenic DI) is the most common type and is caused by lack of vasopressin (ADH) production by a diseased or destroyed posterior pituitary gland. Lack of ADH results in massive diuresis, because ADH normally prompts the kidney to concentrate the urine. Approximately 50% of central DI is idiopathic, as diagnostic testing does not reveal a cause. Central DI is usually permanent, but the signs and symptoms (i.e., thirst, drinking fluids, and urination) are controlled by daily use of synthetic vasopressin.
• Nephrogenic DI (NDI) is caused by inability of the kidneys to respond to normal amounts of ADH, resulting from a variety of drugs or kidney diseases including genetic predisposition. The collecting tubules have decreased permeability to water caused by decreased response to vasopressin by the nephrons. NDI does not improve with synthetic vasopressin and may not improve when probable causes are managed. Familial NDI requires lifelong management. Treatments partially relieve the signs and symptoms. Medications, including lithium, amphotericin B, and demeclocycline can induce NDI. Hypercalcemia can sometimes prompt NDI.
• Gestational or gestogenic DI results from a lack of vasopressin that develops during the third trimester of pregnancy if the pregnant woman’s thirst center is abnormal, causing a blunted thirst response, and/or the placenta destroys vasopressin too rapidly. The placenta may increase the action of vasopressinase, the enzyme that breaks down vasopressin. The condition is controlled using synthetic vasopressin until the DI resolves. Vasopressin can generally be discontinued 4 to 6 weeks after delivery. Signs and symptoms of DI will recur with subsequent pregnancies.
• Dipsogenic DI or primary polydipsia results from vasopressin suppression caused by excessive fluid intake. Primary polydipsia is most often caused by an abnormality in the thirst center of the brain. Unquenchable thirst results in water intoxication. Dipsogenic DI is differentiated from central (pituitary) DI using the water deprivation test. There is no cure for dipsogenic DI at present, but symptoms can be safely relieved. Psychogenic polydipsia is another subtype due to psychosomatic causes that has no treatment that is recognized as consistently effective.
In normal individuals, a more concentrated circulating volume stimulates ADH release through activation of osmoreceptors that monitor serum osmolality. ADH is also released as part of the renin-angiotensin-aldosterone mechanism as a result of hypotension sensed by the juxtomedullary apparatus located outside the glomerulus of the kidney. A 5% to 10% decrease in arterial BP is necessary to increase circulating vasopressin concentrations. Progressive hypotension in healthy individuals results in an exponential increase in plasma ADH via baroreceptor stimulation, while osmoregulated ADH release in response to dehydration is more linear. If the hypothalamus is damaged, production of ADH may not be possible and both the ability to regulate circulating volume and vascular tone may be affected.
8-2 RESEARCH BRIEF
From Wenzel V, Krismer AC, Arntz HR, Sitter H, et al: A comparison of vasopressin and epinephrine for out of hospital cardiopulmonary resuscitation. N Engl J Med 350(2):105–113, 2004.
Endocrine assessment: diabetes insipidus
Goal of assessment
The clinical presentation of DI is dependent on the overall health of the patient and the primary cause. Evaluate for degree of dehydration and its effects on overall hemodynamics, heart rate/rhythm, and mental status. Generally, DI is recognized and managed prior to resulting in serious complications. Practitioners working with brain dead organ donors must be particularly alert to the occurrence of DI. Severe, unmanaged or under managed dehydration in cases of DI may cause hemoconcentration, which may predispose the patient to thrombosis. Dehydration and electrolyte imbalance must be prevented or managed immediately to avoid possible organ damage.
Observation
• Polyuria with dilute urine; 3-20 L/day of urine may be excreted, with specific gravity of 1.000 to 1.005.
• History may include nocturia (getting up frequently at night to urinate) and/or enuresis (bed wetting).
• Dehydration: Poor skin turgor, dry mucous membranes, sunken eyes, slow capillary refill
• Altered mental status: Serum hyperosmolality and hypernatremia affect consciousness and behavior. Changes may also be related to the underlying disease.
Screening labwork
• Point-of-care (POC) capillary blood glucose to rule out hyperglycemia as the cause for diuresis
• Fluid and electrolyte imbalance screening:
Differential Diagnosis of Diabetes Insipidus
| Test | Purpose | Abnormal Findings |
|---|---|---|
| Urine osmolality | Assesses for decreased concentration or dilute urine | Decreased to <200 mOsm/kg; may be higher if volume depletion is present |
| Urine specific gravity | Assesses for dilute urine | Specific gravity: <1.005 Normal is 1.010–1.025 |
| Serum osmolality | Assesses for concentrated blood/hemoconcentration | Increased to >290 mOsm/kg |
| Serum sodium | Monitors for hypernatremia | Increased to >147 mEq/L |
| Plasma ADH level (vasopressin level) | Assesses if vasopressin is elevated or decreased | Central DI: Decreased Nephrogenic DI: Normal or increased Gestational DI: Decreased Dipsogenic DI: Decreased |
| Water deprivation test (Miller-Moses Test) Dehydration should prompt the kidneys to concentrate urine. | To distinguish between the types of DI. Assesses for changes in weight, serum and urine osmolality, and specific gravity when fluid intake is prohibited. | Differentiates psychogenic polydipsia from DI. Central DI and NDI are unaffected by this test. |
| ADH (Vasopressin) test ADH administration will correct the problem if ADH was lacking. | Assesses if the kidneys begin to concentrate urine when ADH is administered. Distinguishes NDI from other types of DI. | Corrects central/neurogenic DI, wherein ADH is lacking. NDI is unaffected by ADH administration, since the problem is unrelated to lack of ADH. |
| Brain or Pituitary magnetic resonance imaging (MRI) | MRI scan used to identify pituitary lesions that may have caused the DI. | If the patient has the “bright spot” or hyperintense emission from the posterior pituitary gland, the patient likely has primary polydipsia. If the “bright spot” is small or absent, the patient likely has central DI. |
Collaborative management
Care priorities
1. Rehydrate if dehydration and/or hypovolemia are present
• Place at least two large-bore IV lines, or have a central line inserted.
• Administer hypotonic IV fluids (5% Dextrose solution, 0.45% saline.). Hyperglycemia and volume overload should be avoided.
• Avoid overly aggressive correction of hypernatremia. Do not decrease sodium level by more than 5mEq/L per hour or 12 mEq/L within 24 hours.
• Patient may be allowed to drink fluids, with the volume accurately recorded
• Monitor urine output judiciously. Total water deficit may be estimated by assuming body water composes approximately 60% of total body weight in kg.
• Monitor continuous ECG for tachycardia and dysrhythmias.
• Evaluate basic ABCs: airway, breathing, and circulation if the patient becomes hypotensive.
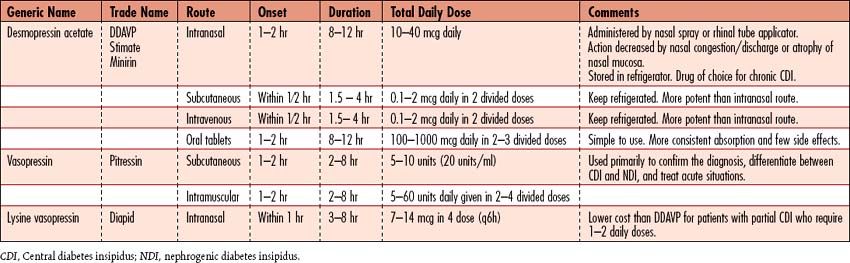
2. Administer exogenous adh (vasopressin)
• Use of desmopressin acetate (DDAVP) is popular because it produces fewer side effects (Table 8-2).
• Several preparations are available, and dosage is adjusted to patient response to DI management. Vasopressin’s potential vasoconstrictive effects occur rarely with appropriate dosing for management of DI. Excessive dosing of ADH may cause hypertension and cardiac symptoms; other side effects include abdominal cramping and increased peristalsis.
5. Manage ndi (adh insensitive) with pharmacotherapy
• Thiazide diuretics (e.g., hydrochlorothiazide [HCTZ]) in combination with a low-sodium diet are the major form of therapy for NDI, to reduce the loss of free water in the urine.
• Chlorpropamide stimulates the release of ADH and facilitates the renal response to ADH.
• Amiloride hydrochloride (a potassium-sparing diuretic) is the medication of choice for the treatment of lithium-induced NDI.
• Nonsteroidal anti-inflammatory drugs (NSAIDs) such as indomethacin have been used as adjunctive therapy in NDI.
CARE PLANS FOR DIABETES INSIPIDUS
related to diuresis secondary to ADH deficiency or altered ADH action
![]()
Fluid Balance, Electrolyte and Acid-Base Balance, Hydration
1. Monitor vital signs every 15 minutes until patient is stable for 1 hour. Monitor CVP, MAP, and, if hemodynamic monitoring was in place, pulmonary artery pressure (PAP), and pulmonary capillary wedge pressure (PCWP), if ordered. Consult physician or midlevel practitioner for the following: HR greater than 140 bpm or BP less than 90/60 or decreased 20 mm Hg or greater, or MAP decreased 10 mm Hg or greater from baseline, CVP less than 2 mm Hg, and PAWP less than 6 mm Hg. Manage judiciously in all patients, including brain-dead organ donors.
2. ![]() Monitor hydration status: mucous membranes, pulse rate and quality, and BP. Excessive water intake may result in fluid overload, particularly in elders and children.
Monitor hydration status: mucous membranes, pulse rate and quality, and BP. Excessive water intake may result in fluid overload, particularly in elders and children.
3. Administer hypotonic solutions (e.g., D5W, D50.25, or 0.45 NaCl) for intracellular rehydration. Usually, fluids are administered as follows: 1 ml IV fluid for each 1 ml of urine output. In patients with brain injury, moderate diuresis may be permitted to avoid the need for administering osmotic diuretics. Hypernatremia, if present, must be corrected slowly (at a rate no greater than 0.5 mEq/L/hr or 12 mEq/L/day) to prevent cerebral edema, seizures, permanent neurologic damage, or death.
4. Administer vasopressin (DDAVP) as ordered. Observe for and document effects. Also be alert to side effects of therapy: hypertension, cardiac ischemia, and hyponatremia.
5. Weigh patient daily, at the same time and using the same scale and garments to prevent error. Consult physician or midlevel practitioner for weight loss greater than 1 kg/day.
6. Observe for indications of dehydration (e.g., poor skin turgor, delayed capillary refill, weak/thready pulse, dry mucous membranes, hypotension).
7. ![]() Monitor for fluid overload, which can occur as a result of rapid infusion of fluid or excessive fluid intake in patients with heart failure: jugular vein distention, dyspnea, crackles (rales), and CVP greater than 12 mm Hg.
Monitor for fluid overload, which can occur as a result of rapid infusion of fluid or excessive fluid intake in patients with heart failure: jugular vein distention, dyspnea, crackles (rales), and CVP greater than 12 mm Hg.
8. If urinary catheter has been removed, observe for resolution of nocturia (waking up at night to urinate) and enuresis (bed wetting) as treatment progresses.
1. Monitor laboratory studies, observing for an appropriate response to treatment, including a decrease in serum sodium, increase in serum and urine osmolality, and increase in urine specific gravity.
2. Monitor urine specific gravity hourly to evaluate response to therapy. Patients may be allowed to develop hypotonic polyuria between doses of vasopressin to demonstrate persistence of DI when transient DI is suspected.
3. Report lack of improvement or deterioration to the physician or midlevel practitioner. Urine output greater than 200 ml/hr for 2 consecutive hours, or 500 ml/hrs in the presence of risk factors should be reported.
4. Instruct patients with permanent DI to wear a medical-alert bracelet labeled with DI. Immediate family members should be familiar with the patient’s current treatment plan in case they are contacted in an emergency.
Disturbed sensory perception (visual and auditory)
resulting from hyperosmolality, dehydration, or hypernatremia
![]()
1. Monitor neurologic status frequently. Notify physician or midlevel practitioner of deterioration.
2. Keep bed in lowest position with side rails raised, if patient is confused.
3. Consider nasogastric tube with suction for comatose or brain-dead organ donor patients to decrease likelihood of aspiration.
4. Elevate head of the bed (HOB) to 30 degrees to minimize the risk of aspiration.
![]()
Infection Severity, Immune Status
1. For patients who have undergone transphenoidal hypophysectomy, inspect nasal packing often for frank bleeding or evidence of CSF leak. If glucose is detected in clear nasal drainage (tested using a glucose reagent stick), CSF is leaking, which indicates a flaw in cranial bone integrity. (See Care of the Patient After Intracranial Surgery, p 638.)
2. Elevate the HOB to minimize the chance of bacterial migration into the brain if CSF leak is suspected. Consult physician or midlevel practitioner promptly.
3. Monitor for infection, including elevated temperature, nuchal rigidity, and altered LOC.
4. Monitor for increased WBC count, which initially may reflect dehydration or the stress response.
5. Since patient is at higher risk for bacterial infection, invasive lines should be managed carefully to avoid bloodstream infection (BSI). Central lines should be removed as soon as possible.
6. To prevent injury and contamination of operative site, patients should not brush their teeth until instructed to do so by physician. Provide sponge-tipped applicators for oral hygiene.
![]() Incision Site Care; Infection Protection; Neurologic Monitoring
Incision Site Care; Infection Protection; Neurologic Monitoring
Deficient knowledge: illness care
![]()
Knowledge: Illness Care; Knowledge: Medication; Knowledge: Treatment Regimen
1. Teach patient appropriate administration of exogenous vasopressin and its side effects.
2. Explain exogenous hormone replacement if the anterior pituitary gland was damaged or removed during surgery. If patient is also experiencing anterior pituitary dysfunction (panhypopituitarism), teach the indicators of hormone replacement excess or deficiency.
3. Demonstrate the method for accurate measurement of urine specific gravity and the importance of keeping accurate records of test results.
4. Teach when to seek medical attention, including signs of dehydration (hypernatremia) and water intoxication (hyponatremia).
5. Explain the importance of obtaining a medical-alert bracelet and identification (ID) card.
6. Stress the importance of continued medical follow-up.
7. For patients with permanent need for hormone replacement, explain the method for obtaining a medical-alert bracelet and ID card outlining diagnosis and appropriate treatment in the event of an emergency.
![]() Teaching: Disease Process; Teaching: Prescribed Medication; Emotional Support
Teaching: Disease Process; Teaching: Prescribed Medication; Emotional Support
Additional nursing diagnoses
If patient has developed DI following a transphenoidal hypophysectomy, see Decreased intracranial adaptive capacity, Ineffective breathing pattern, Risk for infection, and Pain in Care of the Patient After Intracranial Surgery, p. 638; also see Hypernatremia and Hyponatremia in Fluid and Electrolyte Disturbances, p 37.)
Hyperglycemia
Pathophysiology
Patients in the Van den Berghe study who received tight glycemic control for new-onset hyperglycemia realized a much greater improvement in outcomes than did patients with diabetes mellitus, a particularly difficult concept for the medical community to accept. A subset of those with “new-onset” hyperglycemia were found to be undiagnosed diabetics, which prompted a recommendation that all patients undergo glycohemoglobin screening (hemoglobin A1c) performed on hospital admission. Assessment of glycemic control prior to hospitalization helps care providers anticipate whether patients will need insulin following hospitalization. Those with diabetes mellitus may be discharged on oral hypoglycemic agents or insulin. “Stress responders” without diabetes generally do not require insulin following hospitalization, because the stress of their illness or procedure resolves.
8-3 RESEARCH BRIEF
From Van den Berghe G, et al: Intensive insulin therapy in critically ill patients. N Engl J Med 345(19):1359–1367, 2001.
Physicians create insulin titration regimens, because no one method of insulin dosing has been universally accepted. Unlike the frequent titration of medications used to manage hypotension or hypertension, most dosage adjustments of IV insulin infusions are done hourly, because a continuous reading of glucose level is not possible. Dose responses are more difficult to assess. Few protocols individualize dosing based on insulin sensitivity or resistance. Individualization increases the complexity of care if nurses must perform mathematical calculations to make dosing adjustments.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


