Chapter 22 Endocrine complications of HIV infection
The Thyroid
Thyroid pathology
Clinically significant OIs of the thyroid occur very rarely in patients with AIDS, and the advent of ART has further reduced the frequency. In an autopsy study, two-thirds of thyroids examined demonstrated pathologic changes, though no patients had pre-mortem clinical thyroid disease. Indeed, more than one OI was found in the majority of specimens [1]. Pathogens infecting the thyroid included Pneumocystis jiroveci, cytomegalovirus (CMV), Cryptococcus neoformans, Aspergillus fumigatus, Rhodococcus equi, Haemophilus influenzae, Microsporidia, Histoplasma capsulatum, Paracoccidioides brasiliensis, Mycobacterium avium intracellulare, and Mycobacterium tuberculosis. Pneumocystis jiroveci was the most common OI of the thyroid, occurring primarily among patients receiving aerosolized pentamidine [2]. Clinical manifestations of thyroiditis are variable and may include signs and symptoms of either hyper- or hypothyroidism. Palpation of the thyroid gland may reveal localized tenderness and/or fluctuance; systemic signs of infection may be present. Functional testing may reveal transient hyperthyroidism (that may not require treatment), euthyroidism, or hypothyroidism. HIV has also been isolated from thyroid tissue of infected patients with Graves’ disease (GD), though it is unclear whether HIV is involved in the pathogenesis of this autoimmune thyroid disorder [3].
Neoplastic infiltration of the thyroid is uncommon in patients with AIDS. Only two cases of thyroid lymphoma, one case of thyroid carcinoma, and one case of acute myelogenous leukemia presenting as a thyroid mass [4] have been reported. There have been a few cases of Kaposi’s sarcoma (KS) involving the thyroid gland in patients with pre-existing cutaneous lesions. Other frequent pathologic changes of the thyroid include non-specific focal chronic inflammation, colloid goiter, and lipomatosis [1].
Alterations in thyroid function
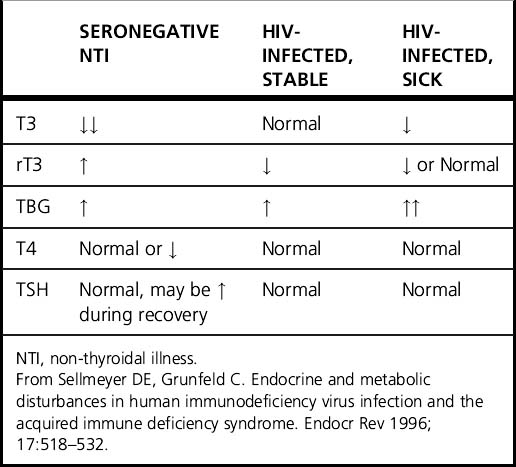
Although asymptomatic HIV-infected patients with stable body weight are usually clinically euthyroid, abnormal thyroid homeostasis is often present in patients with AIDS. In non-thyroidal illness (NTI), inhibition of peripheral T4 to T3 conversion and reduced reverse T3 (rT3) clearance results in low T3 and elevated rT3, usually accompanied by normal thyroid-stimulating hormone (TSH) levels. The characteristic changes of NTI differ from those observed in AIDS patients, who often have higher T3 levels and lower rT3 levels than would generally be expected. The increased thyroid hormone-binding globulin (TBG) levels associated with HIV infection do not explain these findings [5]. The abnormalities of thyroid testing in HIV-infected individuals compared with seronegative patients with NTI are presented in Table 22.1. The maintenance of normal T3 levels among AIDS patients has led to concern that the protective effects that reduction of T3 provides in NTI, such as lowering metabolic rate, ameliorating weight loss, and decreasing protein catabolism, may be compromised. Lower T3 levels are observed, however, during secondary infection and anorexia. Patients with asymptomatic HIV infection have been reported to demonstrate characteristics of compensated hypothyroidism including high-normal range 24-h TSH profiles, lower free T4 levels, and a greater TSH response to TRH infusion [6]. These alterations in thyroid hormone physiology may be adaptive to chronic illness, decreased energy intake, or the increased metabolic demands of HIV infection. Still, there is little evidence to suggest those with asymptomatic HIV infection be routinely screened for thyroid dysfunction [7]. If hyper- or hypothyroidism are clinically suspected, a physical examination that includes an assessment of the thyroid gland should be performed followed by measurement of TSH level. HIV-infected patients with an acute illness, such as local or systemic infection, may have thyroid function test (TFT) results consistent with NTI. In this instance, TFTs should be repeated a few weeks after recovery from illness to document resolution of abnormalities. Use of exogenous thyroid hormone is not indicated in the setting of NTI.
Effects of antiretroviral and other therapies
Rifampin, ketoconazole, and ritonavir may alter thyroid function by accelerating the metabolic clearance of thyroid hormone and can precipitate hypothyroidism in patients with marginal thyroid reserve; thus, higher doses of thyroxine may be necessary in patients receiving concomitant replacement therapy [2].
Treatment of hepatitis C infection with interferon-α (INF-α) has been associated with the development of autoimmune thyroid diseases (AITD) such as GD, Hashimoto’s thyroiditis, and subacute or destructive thyroiditis. Since individuals with pre-existing or recently detected thyroid autoantibodies (i.e. thyroid peroxidase [TPOAb] and thyroglobulin [TgAb] antibodies) are at higher risk for INF-α-induced AITD, practitioners should test TSH, free T4, and thyroid antibodies before initiation of INF-α treatment, followed by measurement of TSH every 8–12 weeks during treatment [8]. IFN-α treatment should be delayed until correction of pre-existing thyroid dysfunction. If thyroid dysfunction develops during treatment, IFN-α need not be discontinued unless destructive thyroiditis with severe symptoms refractory to β-blockers or GD requiring high doses of anti-thyroidal medications develops. Although most patients who develop thyroid dysfunction during treatment with INF-α normalize after it is discontinued, a minority continue to require treatment [8].
Since the introduction of HAART, AITD has been reported as a late complication of immune reconstitution, typically presenting as GD 1–2 years after initiation of therapy [9]. The onset of AITD is temporally consistent with thymic production of naïve CD4 T cells (the “late” phase of T cell repopulation), and immune dysregulation in those with genetic predisposition may result in thyroid-specific autoimmunity.
The Adrenal
Adrenal pathology
Although pathologic involvement of the adrenal gland was frequently noted during autopsy in the pre-HAART era, clinical adrenal insufficiency (AI) was relatively rare. This is likely explained by the fact that greater than 90% of the adrenal cortices must be destroyed for AI to ensue. Cytomegalovirus adrenalitis was the most common finding. Mycobacterium tuberculosis, M. avium intracellulare, C. neoformans, and Toxoplasma gondii also infect the adrenal gland. Infiltration with KS or lymphoma, hemorrhage, fibrosis, infarction, and focal necrosis were also reported [2].
Alterations in adrenal function
HIV-infected individuals may demonstrate changes in steroid metabolism, including an elevation in basal cortisol levels that may be accompanied by decreased responsiveness to ACTH stimulation. Lower levels of ACTH and the weak adrenal androgen dehydroepiandrosterone (DHEA) are often observed, along with impaired adrenal reserve of the 17-deoxysteroids (corticosterone, deoxycorticosterone, and 18-OH-deoxycorticosterone) [10]. Factors such as cytokines, acting independently of the pituitary gland, may directly enhance cortisol biosynthesis in the absence of an increase in ACTH. In some HIV-infected patients, however, the combination of increased cortisol and ACTH levels suggests hypothalamic activation, although those with late-stage HIV disease often have an attenuated pituitary–adrenal response to corticotropin-releasing hormone (CRH). Compensatory rises in ACTH levels may also develop in those with subclinical AI due to physiologic hormonal feedback mechanisms. In AIDS patients who present with elevated levels of both cortisol and ACTH but manifest paradoxical Addisonian features, peripheral glucocorticoid resistance may be present [11]. Levels of DHEA decline with advancing age and chronic illness, in contrast to levels of cortisol, which remain relatively stable. Interest in DHEA therapy among the HIV community was motivated by studies showing that DHEA inhibits HIV-1 replication and activation in vitro. Cross-sectional studies have associated low DHEA levels and elevated cortisol/DHEA ratios with advanced HIV infection, and low serum concentrations of DHEA have been significantly correlated with CD4 T cell count, weight loss, and progression to AIDS. Small placebo-controlled trials of oral administration of DHEA showed improved quality of life in patients with AIDS without change in CD4 T cell count [12]. However, a recent 6-month randomized, double-blind, placebo-controlled study of 40 HIV-infected subjects with suppressed viral loads showed no changes in immune parameters, lean muscle mass, or bone density following treatment with oral DHEA [13]. DHEA cannot be recommended in the routine treatment of HIV-infected patients until its efficacy has been proven in larger, randomized clinical trials evaluating multiple outcomes.
Effects of antiretroviral and other therapies
Although the dorsocervical fat pad enlargement and visceral adiposity seen in some HIV-infected patients appears phenotypically similar to Cushing’s syndrome, overt hypercortisolism has not been found in affected patients [14]. The demonstration of both normal diurnal cortisol excretion and normal response to exogenous CRH administration provides additional evidence that the development of central lipohypertrophy cannot be attributed to abnormal cortisol metabolism, though some have hypothesized that it may be related to the increased cortisol/DHEA ratio observed in these patients. Iatrogenic Cushing’s syndrome, however, can occur in patients treated concomitantly with ritonavir and nasal or inhaled fluticasone (for allergic rhinitis or asthma). Ritonavir prolongs the half-life of fluticasone via effects on CYP3A4, leading to much higher plasma levels of fluticasone than pharmacologically intended and classic physical manifestations of glucocorticoid excess. When given in conjunction with ritonavir, intra-articular triamcinolone used in the treatment of osteoarthritis may also be associated with signs and symptoms of glucocorticoid excess, though the mechanism for this is unclear. Upon steroid withdrawal, secondary adrenal suppression may ensue. Associated conditions such as osteoporosis or diabetes may be induced or exacerbated. Practitioners should be aware of these potential interactions in order to avoid delays in diagnosis among patients who have pre-existing central lipohypertrophy that might mask the clinical features of Cushing’s syndrome. These patients should be examined for cardinal signs of Cushing’s syndrome, including violaceous striae, bruising, and proximal muscle weakness.
Multiple medications used to treat complications of HIV may affect adrenocortical function. AI may be induced in patients with impaired adrenal reserve by conazoles (keto-, flu- and itraconazole) through inhibition of cortisol biosynthesis. Rifampin may also induce AI by increasing the metabolic clearance of cortisol. A syndrome of mineralocorticoid excess has been reported in patients on high-dose itraconazole [15]. Megestrol acetate, a progestational agent used as an appetite stimulant in the treatment of AIDS-wasting, can suppress both the hypothalamic–pituitary–adrenal (HPA) axis and the hypothalamic–pituitary–gonadal (HPG) axis due to its intrinsic glucocorticoid-like activity. Some patients receiving long-term therapy may develop iatrogenic Cushing’s syndrome and/or diabetes mellitus, as well as adrenal failure when treatment is suddenly discontinued. Opiate use, often seen coexisting with HIV infection, can blunt cortisol secretion in response to ACTH stimulation [16].
Treatment considerations
Patients with documented AI should be treated with glucocorticoid replacement therapy and require increased doses during periods of stress. If primary adrenal failure is present with evidence of concomitant mineralocorticoid deficiency (hyperkalemia and metabolic acidosis), the addition of fludrocortisone should be considered. Controversy exists regarding whether patients with elevated basal cortisol levels, but a blunted response to standard single-dose ACTH stimulation, should be treated with glucocorticoid therapy. Some of these patients probably do not require chronic glucocorticoid replacement, as they show adequate cortisol response after receiving supraphysiologic ACTH stimulation for 3 consecutive days [10]. These challenging cases must be evaluated individually, with the goal of minimizing unnecessary glucocorticoid exposure. Administration of short-term, supplementary glucocorticoids to symptomatic patients who demonstrate a subnormal rise in cortisol levels during periods of stress is reasonable.
The Pancreas
Pancreatic pathology
Morphologic abnormalities of the pancreas are common at autopsy in AIDS patients (up to 90%); however, most lesions are asymptomatic [17]. Pancreatic OIs such as mycobacteria, toxoplasmosis, CMV, and P. jiroveci have been documented, with presentation similar to that of pancreatitis due to other causes [18]. The most common pancreatic OI is tuberculosis, presenting with diverse manifestations, such as masses mimicking carcinoma, obstructive jaundice, pancreatitis, gastrointestinal bleeding, and generalized lymphadenopathy. It may be diagnosed by abdominal computed tomography followed by FNA biopsy. HIV-associated neoplasms rarely affect the pancreas, although primary pancreatic lymphoma [19] and KS have been reported, the latter successfully treated with intensive ART and paclitaxel [20].
Alterations in glucose homeostasis and effects of antiretroviral therapy
Prior to the advent of HAART, symptomatic HIV-infected men were found to have increased insulin sensitivity of peripheral tissues compared with non-infected controls. Recent studies found conflicting evidence as to whether insulin sensitivity is altered among asymptomatic HIV-infected individuals who are ARV-naïve. Following the introduction of HAART with protease inhibitors (PIs), abnormalities of glucose metabolism including insulin resistance, impaired insulin secretion, hyperglycemia, and frank diabetes were reported. However, it was unclear whether restoration of health, immune reconstitution, body composition changes, or other antiretrovirals contributed to these disturbances. Subsequent studies of individual PIs among healthy, HIV-seronegative volunteers minimized confounding by HIV-related factors and demonstrated a spectrum of effects of PIs on glucose metabolism [21–28]. For instance, although a single dose of indinavir and ritonavir-boosted lopinavir were found to acutely induce insulin resistance, amprenavir and boosting-dose ritonavir did not. After 4 weeks of treatment, insulin resistance and increased fasting glucose occurred with indinavir but not with ritonavir-boosted lopinavir. Endogenous glucose production (EGP) was increased following administration of indinavir compared to placebo. This finding, indicating an indinavir-induced reduction in the ability of insulin to blunt EGP that may lead to hyperglycemia and a predisposition to diabetes, was not shown with amprenavir. More recently, though it was initially reported that PIs as a class impair pancreatic β-cell insulin secretion in HIV-infected patients, there was no effect on insulin secretion after healthy HIV-seronegative volunteers were given ritonavir-boosted lopinavir for 4 weeks. Thus, PIs do not have a singular class effect on glucose metabolic pathways, and the distinct effects of individual PIs on glucose metabolism must be taken into account when tailoring antiretroviral regimens.
Effects of other therapies
Pentamidine, used in the prevention and treatment of P. jiroveci, may cause pancreatic β-cell toxicity when administered either intravenously or aerosolized. Acute insulin secretion and resultant hypoglycemia may be followed by β-cell destruction and diabetes mellitus [2]. Acute pancreatitis rarely occurs with pentamidine, trimethoprim-sulfamethoxazole, the NRTIs ddI and ddC, ritonavir-induced hypertriglyceridemia and antifungal treatment with liposomal amphotericin B, micafungin, griseofulvin, fluconazole, itraconazole, and voriconazole [15].
The intrinsic glucocorticoid-like activity of megestrol acetate may exacerbate or cause hyperglycemia, although the incidence of this complication is low. Growth hormone (GH) can induce insulin resistance and predispose to hyperglycemia and diabetes. A small increase in hemoglobin A1c accompanies tesamorelin treatment to reduce visceral adipose tissue. Ketoconazole, fluconazole, and voriconazole have been associated with hypoglycemia, though the underlying mechanisms are unknown [15]. Additionally, co-administration of fluconazole with the oral hypoglycemic medications tolbutamide, glimepiride, and nateglinide increase their peak plasma concentrations, increasing risk of hypoglycemia.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


