(eck-you-LIZ-you-mab)
Soliris
Monoclonal antibody
Complement inhibitor
pH 7
Usual dose
A meningococcal vaccine must be administered at least 2 weeks before initial dosing with eculizumab to all patients who have not been previously vaccinated. A booster dose may be required for patients previously vaccinated. Revaccinate according to current medical guidelines. Quadrivalent, conjugated meningococcal vaccines are strongly recommended.
Paroxysmal nocturnal hemoglobinuria (PNH):
Administer 600 mg as an infusion weekly for the first 4 weeks, followed by 900 mg for the fifth dose 1 week later, then 900 mg every 2 weeks thereafter.
Atypical hemolytic uremic syndrome (aHUS) in patients 18 years of age or older:
Administer 900 mg as an infusion weekly for the first 4 weeks, followed by 1,200 mg for the fifth dose 1 week later, then 1,200 mg every 2 weeks thereafter.
Pediatric dose
See general comments under Usual Dose.
Atypical hemolytic uremic syndrome (aHUS) in patients less than 18 years of age:
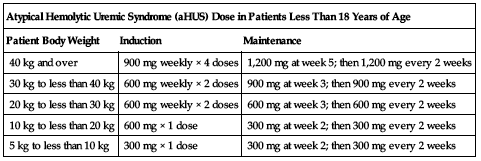
Administer eculizumab based on body weight as outlined in the following chart.
| Atypical Hemolytic Uremic Syndrome (aHUS) Dose in Patients Less Than 18 Years of Age | ||
| Patient Body Weight | Induction | Maintenance |
| 40 kg and over | 900 mg weekly × 4 doses | 1,200 mg at week 5; then 1,200 mg every 2 weeks |
| 30 kg to less than 40 kg | 600 mg weekly × 2 doses | 900 mg at week 3; then 900 mg every 2 weeks |
| 20 kg to less than 30 kg | 600 mg weekly × 2 doses | 600 mg at week 3; then 600 mg every 2 weeks |
| 10 kg to less than 20 kg | 600 mg × 1 dose | 300 mg at week 2; then 300 mg every 2 weeks |
| 5 kg to less than 10 kg | 300 mg × 1 dose | 300 mg at week 2; then 300 mg every 2 weeks |
Dose adjustments
A variance of 1 to 2 days in the scheduled administration time points is allowed if indicated. ■ Age, gender, race, and renal function do not appear to influence the pharmacokinetics of eculizumab. ■ Supplemental dosing of eculizumab is required in the setting of concomitant support with PE/PI (plasmapheresis or plasma exchange, or fresh frozen plasma infusion). See the following chart for supplemental dosing based on the type of intervention.
| Supplemental Dose of Eculizumab After PE/PI | |||
| Type of Intervention | Most Recent Eculizumab Dose | Supplemental Eculizumab Dose with Each PE/PI Intervention | Timing of Supplemental Dose |
| Plasmapheresis or plasma exchange | 300 mg | 300 mg per each plasmapheresis or plasma exchange session | Within 60 minutes after each plasmapheresis or plasma exchange |
| Plasmapheresis or plasma exchange | 600 mg or more | 600 mg per each plasmapheresis or plasma exchange session | Within 60 minutes after each plasmapheresis or plasma exchange |
| Fresh frozen plasma (FFP) infusion | 300 mg or more | 300 mg per each infusion of fresh frozen plasma | 60 minutes before each infusion of fresh frozen plasma |

Dilution
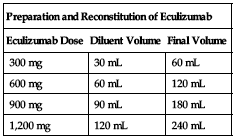
Available in 300 mg/30 mL single-use vials (10 mg/mL). Withdraw the required dose of eculizumab (2 vials are required for the 600-mg dose, 3 vials are required for the 900-mg dose, and 4 vials are required for the 1,200-mg dose) and transfer it to an infusion bag. Must be further diluted to a 5 mg/mL concentration by adding an amount of NS, 1/2NS, D5W, or Ringer’s lactate to the infusion bag equal to the total volume of the eculizumab.
| Preparation and Reconstitution of Eculizumab | ||
| Eculizumab Dose | Diluent Volume | Final Volume |
| 300 mg | 30 mL | 60 mL |
| 600 mg | 60 mL | 120 mL |
| 900 mg | 90 mL | 180 mL |
| 1,200 mg | 120 mL | 240 mL |
Invert gently to ensure thorough mixing. Allow the diluted solution to reach room temperature before infusion. Do not use an artificial heat source (e.g., microwave). Discard unused portions; contains no preservatives.
Filters:
Specific information not available.
Storage:
Refrigerate vials in original carton at 2° to 8° C (36° to 46° F) and protect from light. Do not use beyond the expiration date on the vial. Diluted solution is stable for 24 hours refrigerated or at CRT. Do not freeze or shake.
Compatibility
Specific information not available.
Rate of administration
Do not administer as an IV push or a bolus injection. For infusion via gravity feed, syringe-type pump, or infusion pump.
Adults:
A single dose as an infusion over 35 minutes. If infusion is slowed or stopped for any reason, the total infusion time should not exceed 2 hours.
Pediatric patients:
A single dose as an infusion over 1 to 4 hours.
Actions
A recombinant, DNA-derived, humanized IgG monoclonal antibody. A genetic mutation in patients with paroxysmal nocturnal hemoglobinuria (PNH) leads to the generation of abnormal RBCs (known as PNH cells) that are deficient in terminal complement inhibitors; this deficiency makes these RBCs sensitive to persistent terminal complement-mediated destruction. Ongoing destruction of these RBCs is called hemolysis. Eculizumab, a complement inhibitor, specifically binds to the complement protein C5 and prevents complement-mediated intravascular hemolysis. It improves the lives of patients suffering from this disease by directly targeting the underlying disease process and markedly decreasing the ongoing RBC destruction that causes the hemolysis responsible for the S/S of PNH. In aHUS, impairment in the regulation of complement activity leads to uncontrolled terminal complement activation, resulting in platelet activation, endothelial cell damage, and thrombotic microangiopathy. Eculizumab inhibits the complement-mediated thrombotic microangiopathy (TMA) in patients with aHUS. Half-life is 190 to 354 hours.
Indications and uses
A complement inhibitor for the treatment of patients with paroxysmal nocturnal hemoglobinuria (PNH) to reduce hemolysis. PNH is a rare, disabling, and life-threatening genetic mutation blood disorder defined by chronic RBC destruction (hemolysis). Symptoms may include anemia; disabling fatigue; dysphagia; dyspnea; erectile dysfunction; hemoglobinuria; jaundice; recurrent pain in the abdomen, back, or head; renal dysfunction; and thromboses. Average age of onset is the early 30s. ■ Treatment of patients with atypical hemolytic uremic syndrome (aHUS) to inhibit complement-mediated thrombotic microangiography.
Limitation of use:
Eculizumab is NOT indicated for treatment of patients with Shiga toxin E. coli–related hemolytic uremic syndrome (STEC-HUS).
Contraindications
Do not use in patients with unresolved serious Neisseria meningitidis infection or in patients not currently vaccinated against N. meningitidis unless the risk of delaying eculizumab therapy outweighs the risk of developing meningococcal infection.
Precautions
For IV infusion only; do not administer by IV push or bolus injection.
■ Susceptibility to serious meningococcal infections (septicemia and/or meningitis) is increased. Meningococcal infections may become rapidly life threatening or fatal if not recognized and treated early. ■ Comply with the most current Advisory Committee on Immunization Practices (ACIP) recommendations for meningococcal vaccination in patients with complement deficiencies. Revaccinate according to current medical guidelines, considering the duration of eculizumab therapy. ■ A meningococcal vaccine must be administered at least 2 weeks before initial dosing with eculizumab to all patients who have not been previously vaccinated unless the risks of delaying eculizumab therapy outweigh the risks of developing meningococcal infection. If urgent eculizumab therapy is indicated in an unvaccinated patient, administer the meningococcal vaccine as soon as possible. In clinical studies, such patients received antibiotics for prophylaxis of meningococcal infection until at least 2 weeks after vaccination. Benefits and risks of antibiotic prophylaxis not established. Vaccination reduces, but does not eliminate, the risk of meningococcal infections. ■ Use caution in patients with any systemic infection. ■ Eculizumab blocks terminal complement activation. Patients have increased susceptibility to infections, especially with encapsulated bacteria. Aspergillus infections have occurred in immunocompromised and neutropenic patients. Pediatric patients may be at increased risk of developing serious infections due to Streptococcus pneumoniae or Haemophilus influenzae type B (Hib). Administer vaccinations for prevention of these infections according to medical guidelines. ■ Serious hemolysis may occur in patients who discontinue eculizumab therapy; see Monitor. ■ A protein product; infusion reactions may occur. Hypersensitivity reactions, including anaphylaxis, are possible; however, infusion reactions severe enough to discontinue eculizumab did not occur during clinical trials. ■ Has a potential for immunogenicity. Low titers of antibodies to eculizumab have been detected but did not appear to correlate to clinical response. ■ Continue established anticoagulant therapy during eculizumab treatment; the effect of withdrawal of anticoagulant therapy during eculizumab therapy has not been established. ■ Eculizumab is available through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS). Prescribers must be enrolled in the program. Contact manufacturer for further information.
Monitor:
Obtain baseline CBC with differential and platelets, lactic dehydrogenase (LDH), SCr, and bilirubin. ■ LDH levels increase during hemolysis and with TMA. Monitoring may assist in determining the effectiveness of eculizumab therapy. ■ Monitor for early S/S of meningococcal infections (moderate to severe headache with nausea or vomiting, fever, or a stiff neck or stiff back; fever of 103° F [39.4° C] or higher; fever and a rash; confusion; and/or severe muscle aches with flu-like symptoms and light sensitivity). Evaluate immediately and treat with antibiotics if indicated. Discontinue eculizumab during treatment of serious meningococcal infections. ■ Monitor for other types of infections. ■ Monitor for S/S of an infusion or hypersensitivity reaction (e.g., chills, dyspnea, pruritus) during infusion and for at least 1 hour postinfusion. Slow or temporarily discontinue the infusion as indicated. ■ Monitor patients with PNH who discontinue eculizumab for a minimum of 8 weeks to detect serious hemolysis and other reactions (e.g., blood clots; chest pain; confusion; decreased hemoglobin, hematocrit, and haptoglobin; difficulty breathing; elevated LDH, bilirubin, or SCr; free serum hemoglobin; and hemoglobinuria with pink/red urine). ■ Monitor patients with aHUS who discontinue eculizumab for S/S of TMA complications for a minimum of 12 weeks. Clinical S/S of TMA may include angina, dyspnea, mental status changes, seizures, or thrombosis. Changes in laboratory parameters that may indicate TMA include a decrease in platelet count or an increased SCr and LDH.
Patient education:
Read the patient medication guide before initiating eculizumab and before each dose. ■ Meningococcal vaccination is required before initiating therapy. Previously vaccinated individuals may require a booster dose. Vaccination may not prevent meningococcal infection. Important to receive and stay up-to-date on all recommended immunizations. Discuss immunization status with physician. ■ Eculizumab affects the immune system and can lower the ability to fight infections. Immediately report S/S of a meningococcal infection (moderate to severe headache with nausea or vomiting, fever, or a stiff neck or stiff back; fever of 103° F [39.4° C] or higher; fever and a rash; confusion; and/or severe muscle aches with flu-like symptoms and light sensitivity). Manufacturer supplies a patient safety card that lists these symptoms. Card should be carried at all times during treatment and for 3 months after the last dose of eculizumab is administered. Share card with all health care providers treating you. ■ Promptly report other S/S of an infection. ■ Promptly report chills, dyspnea, and/or itching during or soon after an infusion. ■ Report a suspected pregnancy and/or tell your doctor if you are breast-feeding. ■ Stopping the infusions may have serious side effects and requires prolonged monitoring for development of hemolysis or TMA. Increased risk of meningococcal infection continues for several weeks after eculizumab is discontinued.
Maternal/child:
Category C: use during pregnancy only if the benefits justify the potential risk to the fetus. Eculizumab is a recombinant IgG moleculte that is expected to cross the placenta. Pregnant women with PNH and their fetuses have high rates of morbidity and mortality during pregnancy and postpartum. Treatment with eculizumab may increase fetal survival and decrease maternal complications. ■ Use caution if breast-feeding; IgG is secreted in breast milk, but antibodies may not enter the neonatal and infant circulation in substantial amounts. Consider risks to infant versus benefits of breast-feeding. ■ Safety and effectiveness for treatment of PNH in pediatric patients less than 18 years of age not established. ■ Safety and effectiveness for treatment of aHUS appear similar in pediatric and adult patients. Follow medical guidelines for vaccinations for prevention of infections due to Neisseria meningitidis, Streptococcus pneumoniae, and Hib.
Elderly:
Limited experience did not identify age-related differences in safety and effectiveness.
Drug/lab interactions
Formal drug interaction studies have not been completed. ■ Continue established anticoagulant therapy during eculizumab treatment; the effect of withdrawal of anticoagulant therapy during eculizumab therapy has not been established.
Side effects
Meningococcal infections (meningitis and/or septicemia) and the progression of PNH are the most serious side effects reported; may be life threatening and may occur in patients who have been vaccinated. The most commonly reported side effects in patients with PNH are back pain, headache, nasopharyngitis, and nausea. Other reported side effects include constipation, cough, fatigue, herpes simplex infections, influenza-like illness, myalgia, pain in extremity, respiratory tract infection, and sinusitis. The most commonly reported side effects in patients with aHUS are abdominal pain, anemia, cough, diarrhea, fever, headache, hypertension, nasopharyngitis, nausea, peripheral edema, upper respiratory infection, UTI, and vomiting. Other reported side effects include arthralgia, asthenia, back pain, bronchitis, fatigue, gastroenteritis, hypokalemia, hypotension, insomnia, leukopenia, neoplasms (benign, malignant, and unspecified), proteinuria, pruritus, rash, and renal impairment.
Post-marketing:
Cases of serious or fatal meningococcal infections have been reported.
Antidote
Keep physician informed of all side effects. Some will be treated symptomatically. Potential meningococcal infections must be evaluated immediately and treated with antibiotics promptly; may be life threatening. Treat hypersensitivity or infusion reactions as indicated; may respond to slowing or temporarily discontinuing the infusion or may require the use of epinephrine, corticosteroids, diphenhydramine bronchodilators (e.g., albuterol, aminophylline), IV saline, oxygen, and/or acetaminophen. Total infusion time should not exceed 2 hours. If TMA complications occur after eculizumab is discontinued, consider reinstitution of treatment, plasma therapy (plasma exchange or FFP infusion), or appropriate organ-specific supportive measures. Resuscitate as necessary.
Edetate calcium disodium 
(ED-eh-tayt KAL-see-um DYE-so-dee-um)
Calcium Disodium Edetate, Calcium Disodium Versenate, Calcium EDTA
Antidote
Chelating agent
Lead mobilization
pH 6.5 to 8
Usual dose
Specific fluid requirements indicated; see Monitor and Maternal/Child. Do not exceed recommended daily dose.
Asymptomatic adults and pediatric patients with blood lead levels over 20 mcg/dL but under 70 mcg/dL:
1,000 mg/M2/24 hr (50 mg/kg/24 hr) for 3 to 5 days. After a rest period of 2 to 4 days (preferably up to 2 weeks) to allow for redistribution of lead, repeat the process, if indicated, based on severity of lead toxicity and patient tolerance.
Symptomatic adults and pediatric patients with blood levels over 70 mcg/dL:
Dimercaprol (BAL) will be given IM in divided doses every 4 hours for a minimum of 3 or up to 5 days. 4 hours after the first dose of BAL, begin edetate calcium disodium 1,000 mg/M2/24 hr or 25 to 50 mg/kg/24 hr for 5 days. If blood lead concentrations rebound to above 45 mcg/dL within 5 to 7 days after the initial course, repeat the edetate calcium disodium. Do not repeat the dimercaprol regimen.
Dose adjustments
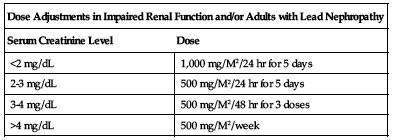
Reduce dose in pre-existing renal disease and/or adults with lead nephropathy. In adults with lead nephropathy dose is based on serum creatinine levels and repeated monthly until lead excretion is reduced toward normal according to the following chart.
| Dose Adjustments in Impaired Renal Function and/or Adults with Lead Nephropathy | |
| Serum Creatinine Level | Dose |
| <2 mg/dL | 1,000 mg/M2/24 hr for 5 days |
| 2-3 mg/dL | 500 mg/M2/24 hr for 5 days |
| 3-4 mg/dL | 500 mg/M2/48 hr for 3 doses |
| >4 mg/dL | 500 mg/M2/week |
Dilution
Add total daily dose to 250 to 500 mL of D5W or NS for infusion.
Storage:
Before use, store at CRT.
Compatibility
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
Manufacturer lists amphotericin B (conventional), hydralazine, D10W, 10% invert sugar in NS, LR, Ringer’s solution, 1/6 M lactate as incompatible. Must be diluted in specific IV solutions; see Dilution.
Rate of administration
References vary greatly. Manufacturer recommends the total daily dose be evenly distributed over 8 to 12 hours. May cause an increase in intracranial pressure with too-rapid injection in patients with lead encephalopathy and cerebral edema.
Actions
A chelating agent. Helps to remove metals, especially lead, from the body. Will form a stable chelate with metals that have the ability to displace calcium from the molecule (e.g., lead, zinc, cadmium). Distributed primarily in the extracellular fluid. Half-life is 20 to 60 minutes. Chelated compounds are excreted in urine; up to 50% in 1 hour and 95% in 24 hours. The primary source of lead chelated by edetate calcium disodium is from bone. Following administration, urinary lead output increases and blood lead concentration decreases, but brain lead is significantly increased due to internal redistribution of lead.
Indications and uses
Reduction of blood levels and depot stores of lead in lead poisoning (acute and chronic) and lead encephalopathy in both pediatric and adult patients.
Unlabeled uses:
Treatment of poisoning by radioactive and nuclear fission products such as plutonium, thorium, uranium, and yttrium. ■ Treatment of poisoning from other heavy metals such as chromium, manganese, nickel, zinc, and possibly vanadium.
Contraindications
Anuria, active renal disease, or hepatitis.
Precautions
Do not confuse with edetate disodium, which does not chelate lead but actually removes calcium from the body and can be very dangerous. ■ Patients with lead encephalopathy and cerebral edema may have a lethal increase in intracranial pressure with IV infusion; IM injection preferred; see Maternal/Child. ■ Equally effective with IM or IV administration. IM route is used for all patients with overt lead encephalopathy and has been suggested as the preferred route by some for young pediatric patients. ■ Usually given IM in pediatric patients, unless given concurrently with BAL (insufficient IM injection sites); see Maternal/Child. ■ May produce toxic and fatal effects. ■ Produces the same renal damage as lead poisoning (e.g., proteinuria and microscopic hematuria). ■ Nephrotoxicity is dose dependent and may be reduced by ensuring adequate diuresis before treatment begins. ■ Use with caution in mild renal disease; see Dose Adjustments. ■ Patients must be removed from the source of contamination promptly. ■ Use for diagnosis of lead poisoning as a lead mobilization test is controversial; see literature. Edetate calcium disodium mobilization test should not be used in symptomatic patients or in patients with blood levels above 55 mcg/dL for whom appropriate therapy is indicated. ■ Not effective in mercury, gold, or arsenic poisoning.
Monitor:
Urine flow must be established before dimercaprol (BAL) or edetate calcium disodium is administered. IV fluids may be used. Avoid excessive fluid in patients with cerebral edema or lead encephalopathy. Once urine flow is established, further IV fluid is restricted in all patients to basal water and electrolyte requirements. ■ Monitor urinalysis, urine sediment, renal and hepatic function, and electrolyte levels before treatment; repeat daily in serious cases and on the second and fifth day in less serious cases. Daily urine specimens are recommended to determine status of renal function. ■ Monitor ECG and vital signs. ■ Elevated erythrocyte protoporphyrin levels (greater than 35 mcg/dL) indicate the need to perform a venous blood lead determination. ■ An elevation of urinary coproporphyrin (greater than 250 mcg/day in adults and greater than 75 mcg/day in pediatric patients under 80 lbs) and an elevation of urinary delta-aminolevulinic acid (greater than 4 mg/day in adults and greater than 3 mg/M2/day in pediatric patients) are associated with blood lead levels greater than 40 mcg/dL. ■ Excretion of calcium is not increased, but excretion of zinc and other essential metals is; monitor and replace as indicated. ■ Obtain specific fluid orders from the physician.
Patient education:
If no urine output for 12 hours, report immediately.
Maternal/child:
Category B: safety for use during pregnancy not established; benefits must outweigh risks. ■ Use caution in nursing mothers. ■ Lead poisoning is often more severe in pediatric patients compared with adult patients. Lead encephalopathy occurs more often in pediatric patients. May be incipient and thus overlooked. Mortality rate in pediatric patients has been high; see Precautions. ■ IV injection has been associated with fatality in some young children, and IM injection is considered to be the preferred route by some clinicians.
Elderly:
Consider age-related organ damage.
Drug/lab interactions
Steroids will increase renal toxicity. ■ Inhibits the action of zinc insulin preparations by chelating the zinc.
Side effects
Acute renal tubular necrosis, anemia, anorexia, arthralgia, cardiac rhythm irregularities, chills, excessive thirst, fatigue, fever, headache, hematuria, hypercalcemia, hypersensitivity (e.g., sneezing, nasal congestion), hypotension, increases in liver function tests (mild), leg and other muscle cramps, malaise, myalgia, nausea, numbness, proteinuria, tetany, tingling, transient bone marrow suppression, tremors, vomiting, weakness, zinc deficiency.
Antidote
Notify the physician of any side effects. Most will improve with a decrease in rate of the infusion or will be treated symptomatically. Discontinue if urine flow stops to avoid high tissue levels of the drug. Discontinue at the first sign of renal toxicity (e.g., presence of large renal epithelial cells or increasing numbers of RBCs). Treat cerebral edema with repeated doses of mannitol. Not known if edetate calcium disodium is dialyzable.
Edetate disodium 
(ED-eh-tayt DYE-so-dee-um)
EDTA Disodium, Endrate
Antihypercalcemic agent
Calcium chelating agent
pH 6.5 to 7.5
Usual dose
50 mg/kg of body weight/24 hr or in equally divided doses every 12 hours (25 mg/kg every 12 hours). Total dose should not exceed 3 Gm/24 hr. Usually given for 5 days, held for 2 days. Regimen may be repeated to a total of 15 doses.
Pediatric dose
40 mg/kg of body weight/24 hr in equally divided doses every 6 to 12 hours (20 mg/kg every 12 hours, 10 mg/kg every 6 hours). Do not exceed 70 mg/kg/24 hr or adult dose, whichever is less. See instructions in Usual Dose.
Dose adjustments
Dose selection should be cautious in the elderly. Reduced doses may be indicated based on potential for decreased organ function and concomitant disease or drug therapy.
Dilution
Recommended dose must be diluted in 500 mL D5W or NS and given as an infusion. A 0.5% solution will reduce the risk of thrombophlebitis. Do not exceed cardiac reserve in any patient. Use less diluent if necessary in pediatric patients. Must be diluted to at least a 3% solution.
Storage:
Store at room temperature.
Compatibility
Specific information not available. Consider specific use; consult pharmacist.
Rate of administration
Must not exceed more than 15 mg of actual medication over 1 minute. Total dose usually given over 3 to 4 hours. Rapid IV infusion may cause a sudden drop in serum calcium, resulting in tetany, convulsions, arrhythmias, and death. Reduce rate and further dilute solution for pain at injection site.
Actions
A calcium-chelating agent. Also forms chelates with other polyvalent metals (e.g., magnesium, zinc). Attracts calcium ions immediately on injection and becomes calcium disodium edetate. Capable of severely depleting the body of calcium stores. Exerts a negative inotropic effect on the heart. It is well distributed in extracellular fluids and rapidly excreted in the urine.
Indications and uses
Treatment of cardiac arrhythmias (atrial and ventricular, especially when caused by digoxin toxicity). ■ Hypercalcemia.
Contraindications
Anuria, known sensitivity to edetate disodium, renal disease.
Precautions
Read label carefully. Deaths have been caused when edetate disodium was administered mistakenly for edetate calcium disodium. ■ Used only when the severity of disease indicates necessity. ■ May produce hypocalcemia quickly, especially if used for purposes other than chelating calcium. ■ Use repeatedly with caution because of potential for nephrotoxicity and mobilization of extracirculatory calcium stores. ■ Use caution in cardiac disease (may adversely affect myocardial contractility), diabetes (lower blood sugar may require less insulin), severe renal disease, liver disease, congestive heart failure (1 Gm of sodium in each 5 Gm), limited cardiac reserve, and patients with a history of seizures or intracranial lesions.
Monitor:
Monitor vital signs and ECG before and during therapy. ■ Confirm patency of vein, avoid extravasation; can cause tissue necrosis. ■ Routine electrolyte panel (potassium deficiency) and urine specimens for casts and cells necessary during therapy. Magnesium, zinc, and other trace element deficiencies can occur. ■ Keep patient in supine position during and after administration (15 to 30 minutes) to avoid postural hypotension. ■ Obtain blood for serum calcium levels just before beginning a new infusion; specific lab methods required. ■ Inhibits coagulation of blood (transient). Liver function tests may be indicated. ■ See Drug/Lab Interactions.
Maternal/child:
Category C: safety for use in pregnancy or breast-feeding not established. Use with extreme caution and only if clearly needed.
Elderly:
Reduced doses may be indicated; see Dose Adjustments. Monitoring of renal function is suggested.
Drug/lab interactions
Inhibits mannitol. ■ Potentiates neuromuscular blocking antibiotics (e.g., gentamicin). ■ Inhibits coagulation of blood (transient). ■ A sudden drop in calcium levels may decrease effects of digoxin. ■ Obtain blood for serum calcium levels just before beginning a new infusion. Specific laboratory methods must be used for accurate evaluation.
Side effects
Anorexia, arthralgia, circumoral paresthesias, diarrhea, fatigue, fever, glycosuria, headache, hyperuricemia, hypotension, malaise, nasal congestion, nausea, numbness, sneezing, tearing, thirst, thrombophlebitis, urinary urgency, vomiting.
Major:
Anaphylaxis, anemia, cardiac arrhythmias, dermatitis, hemorrhage, hypocalcemic tetany, prolonged QT interval, renal tubular destruction (reversible), seizures, death.
Antidote
Notify the physician of any side effect. For progression of minor side effects or any major side effect, discontinue drug immediately and notify the physician. Calcium gluconate is the antidote of choice and should be available for infusion at all times (use extreme caution if patient is digitalized). Treat mild hypotension by maintaining in supine position until recovery. Additional hydration indicated with S/S of nephrotoxicity. Treat anaphylaxis and resuscitate as necessary.
Edrophonium chloride
(ed-roh-FOH-nee-um KLOR-eyed)
Enlon, Tensilon, Tensilon PF
Cholinergic
Cholinesterase inhibitor
Antidote
Diagnostic agent
pH 5.4
Usual dose
1 to 10 mg (0.1 to 1 mL) at specified intervals depending on usage. Maximum dose should never exceed 40 mg (4 doses of 10 mg each).
Myasthenia gravis diagnosis:
10 mg (1 mL) in tuberculin syringe. Give 2 mg (0.2 mL). If no reaction occurs in 45 seconds, give remaining 8 mg (0.8 mL). Test may be repeated after 30 minutes.
Myasthenia treatment evaluation:
1 to 2 mg (0.1 to 0.2 mL) 1 hour after oral intake of drug being used for treatment. Package insert has a chart differentiating myasthenic and nonmyasthenic responses.
Myasthenia crisis evaluation:
2 mg (0.2 mL) in tuberculin syringe. Give 1 mg (0.1 mL). If the patient’s condition does not deteriorate, give 1 mg (0.1 mL) after 60 seconds. Improvement in cardiac status and respiration should occur.
Antagonist to curare and other nondepolarizing muscle relaxants:
10 mg (1 mL). May be repeated as necessary up to 4 doses. (Available in combination with atropine [Enlon-Plus] for use in reversal of nondepolarizing muscle relaxants.)
Terminate paroxysmal atrial tachycardia (unlabeled):
5 to 10 mg as a bolus injection. See Dose Adjustments. Repeat once in 10 minutes if necessary.
Slow supraventricular tachycardias (unlabeled):
2 mg as a test dose. Repeat 2 mg every 1 minute until arrhythmia controlled or total dose of 10 mg is given. If HR decreases, may begin an infusion of 0.25 mg/min. May be increased to 2 mg/min if necessary.
Pediatric dose
May be given IM if the IV route is not available; however, doses are different; check literature. See Maternal/Child.
Myasthenia gravis diagnosis:
Neonates:
0.1 mg (0.01 mL).
Infants:
0.5 mg (0.05 mL).
Pediatric patients less than 34 kg:
1 mg (0.1 mL); if no response in 30 to 45 seconds, give 1 mg (0.1 mL) every 30 to 45 seconds up to 5 mg (0.5 mL). 34 kg or more, give 2 mg (0.2 mL); if no response in 30 to 45 seconds, give 1 mg (0.1 mL) every 30 to 45 seconds up to 10 mg. Another source has the same dose for neonates but recommends 0.2 mg/kg/dose (0.02 mL/kg/dose) for infants and other pediatric patients with 20% of a dose given as a test dose slowly. If no response in 1 minute, give in 1-mg increments to a maximum calculated dose or 10 mg, whichever is less.
Dose adjustments
Reduce antiarrhythmic dose to 5 to 7 mg in the elderly.
Dilution
May be given undiluted. In the treatment of myasthenia crisis, this drug may be diluted in D5W or NS and given as a continuous IV. Use an infusion pump or microdrip (60 gtt/mL).
Filters:
No data available from manufacturer.
Compatibility
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
One source suggests the following compatibilities:
Y-site:
Heparin, hydrocortisone sodium succinate (Solu-Cortef), potassium chloride (KCl).
Rate of administration
2 mg (0.2 mL) or fraction thereof over 15 to 30 seconds.
Curare antagonist:
A single dose over 30 to 45 seconds.
Antiarrhythmic:
See Usual Dose.
Actions
An anticholinesterase and antagonist of nondepolarizing neuromuscular-blocking agents. Inhibits the enzyme acetylcholinesterase, allowing acetylcholine to accumulate at the myoneural junction. Restores normal transmission of nerve impulses. Acts within 30 to 60 seconds and has an extremely short duration of action, seldom exceeding 10 minutes. Produces vagal stimulation, shortens refractory period of atrial muscle, and slows conduction through the AV node.
Indications and uses
Diagnosis of myasthenia gravis. ■ Evaluation of adequate treatment of myasthenia gravis. ■ Evaluation of emergency treatment of myasthenia crisis. ■ An antagonist to nondepolarizing muscle relaxants (e.g., atracurium [Tracrium]). ■ Adjunct in treatment of respiratory depression caused by curare overdosage.
Unlabeled uses:
Termination of supraventricular tachycardia unresponsive to cardiac glycosides. Adenosine is the drug of choice. ■ Diagnosis of supraventricular tachycardia. ■ Evaluate function of a demand pacemaker.
Contraindications
Apnea, known hypersensitivity to anticholinesterase agents, intestinal and urinary obstructions of mechanical type.
Precautions
A physician should be present when this drug is used. ■ The term crisis is used when severe respiratory distress with ventilatory inadequacy occurs. The crisis may be secondary to a sudden increase in severity of myasthenia gravis (myasthenic crisis) or to overtreatment with anticholinesterase drugs (cholinergic crisis). If apnea is present, controlled ventilation must be secured before any testing with edrophonium. ■ Use caution when administering to patients being treated with anticholinesterase drugs (e.g., neostigmine). S/S of cholinergic crisis may mimic those of myasthenic weakness, and the patient’s condition may worsen with administration of edrophonium. ■ Use caution in patients with bronchial asthma, cardiac arrhythmias, or myasthenia gravis treated with anticholinesterase drugs. ■ Isolated cases of respiratory or cardiac arrests have been reported. ■ Contains sulfites; use caution in patients with allergies.
Monitor:
Atropine 1 mg must be available and ready for injection at all times. ■ Continuously observe patient reactions. ■ Anticholinesterase insensitivity may develop; withhold drugs and support respiration as necessary. ■ See Drug/Lab Interactions.
Maternal/child:
Safety for use during pregnancy and breast-feeding not established. Use during pregnancy only if benefit justifies potential risk to mother and fetus. ■ Discontinue breast-feeding. ■ Safety and effectiveness in reversing neuromuscular blockade in pediatric patients not established. However, doses of 0.1 to 1.43 mg/kg have been used; effects (antagonism) were more rapid than in adults.
Drug/lab interactions
Muscarinic effects antagonized by atropine; see Antidote. ■ May be inhibited by corticosteroids and magnesium. ■ May cause bradycardia with digoxin glycosides. ■ Briefly antagonizes the effects of nondepolarizing neuromuscular blocking agents (e.g., atracurium, pancuronium, vecuronium). ■ Prolongs muscle relaxant effect of succinylcholine.
Side effects
Abdominal cramps, anorexia, anxiety, bradycardia, bronchiolar spasm, cardiac arrhythmias and arrest, cold moist skin, contraction of the pupils, convulsions, diarrhea, dysphagia, fainting, increased lacrimation, increased pulmonary secretion, increased salivation, insomnia, irritability, laryngospasm, muscle weakness, nausea, perspiration, ptosis, respiratory arrest (either muscular or central), urinary frequency and incontinence, vomiting.
Antidote
If side effects occur, discontinue the drug and notify the physician. Atropine sulfate in doses of 0.4 to 0.5 mg IV will counteract most side effects and may be repeated every 3 to 10 minutes. Endotracheal intubation or tracheostomy is considered prophylactic in anesthesia or crises. Artificial ventilation, oxygen therapy, cardiac monitoring, adequate suctioning, and treatment of shock or convulsions must be instituted and maintained as necessary. Treat hypersensitivity reactions as indicated.
Elotuzumab
(EL-oh-TOOZ-ue-mab)
Empliciti
Monoclonal antibody
Antineoplastic
Usual dose
Premedication:
To reduce the risk of infusion reactions, premedicate with the following.
Dexamethasone:
Divide into an oral and an IV dose. Administer 28 mg as an oral dose between 3 and 24 hours before elotuzumab infusion. Then 45 to 90 minutes before elotuzumab infusion, administer dexamethasone 8 mg IV, an H1 blocker (diphenhydramine [25 to 50 mg orally or IV] or equivalent), an H2 blocker (ranitidine [50 mg IV or 150 mg orally] or equivalent), and acetaminophen (650 to 1,000 mg orally).
Elotuzumab:
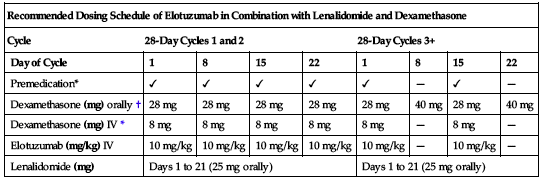
10 mg/kg as an infusion every week for the first 2 cycles and every 2 weeks thereafter in conjunction with the recommended dosing of lenalidomide (Revlimid) and dexamethasone as described in the following chart. Continue treatment until disease progression or unacceptable toxicity. Refer to the prescribing information for dexamethasone and lenalidomide and other premedications as appropriate.
| Recommended Dosing Schedule of Elotuzumab in Combination with Lenalidomide and Dexamethasone | ||||||||
| Cycle | 28-Day Cycles 1 and 2 | 28-Day Cycles 3+ | ||||||
| Day of Cycle | 1 | 8 | 15 | 22 | 1 | 8 | 15 | 22 |
| Premedication* | ✓ | ✓ | ✓ | ✓ | ✓ | — | ✓ | — |
| Dexamethasone (mg) orally† | 28 mg | 28 mg | 28 mg | 28 mg | 28 mg | 40 mg | 28 mg | 40 mg |
| Dexamethasone (mg) IV* | 8 mg | 8 mg | 8 mg | 8 mg | 8 mg | — | 8 mg | — |
| Elotuzumab (mg/kg) IV | 10 mg/kg | 10 mg/kg | 10 mg/kg | 10 mg/kg | 10 mg/kg | — | 10 mg/kg | — |
| Lenalidomide (mg) | Days 1 to 21 (25 mg orally) | Days 1 to 21 (25 mg orally) | ||||||
*Premedicate 45 to 90 minutes before elotuzumab infusion with medications described under Premedication in Usual Dose (dexamethasone IV, an H1 blocker [diphenhydramine IV or PO or equivalent], an H2 blocker [ranitidine IV or PO or equivalent], and acetaminophen PO).
†Give 28 mg of dexamethasone orally between 3 and 24 hours before elotuzumab infusion and 40 mg of dexamethasone orally on days when elotuzumab is NOT administered.
Dose adjustments
If the dose of one drug in the regimen is delayed, interrupted, or discontinued, treatment with the other drugs may continue as scheduled. However, if dexamethasone is delayed or discontinued, base the decision regarding whether to administer elotuzumab on clinical judgment (i.e., risk of hypersensitivity). ■ Interrupt the elotuzumab infusion if a Grade 2 or higher infusion reaction occurs. Upon resolution to Grade 1 or lower, restart elotuzumab infusion at 0.5 mL/min and gradually increase at a rate of 0.5 mL/min every 30 minutes as tolerated to the rate at which the infusion reaction occurred. Resume the escalation regimen if there is no recurrence of the infusion reaction; see Rate of Administration and Antidote. ■ If the infusion reaction recurs, stop the elotuzumab infusion and do not restart on that day. ■ A severe infusion reaction may require permanent discontinuation of elotuzumab. ■ Dose delays and modifications for dexamethasone and lenalidomide should be performed as recommended in their Prescribing Information.
Dilution
Available as a 300-mg or 400-mg single-dose vial. Calculate the dose (mg) based on patient weight, total volume (mL) of elotuzumab solution required, and the number of vials required using the following calculations:
(Weight in kg × dose/kg) ÷ 300 (400) mg/vial = # of vials required
(Weight in kg × dose/kg) ÷ 300 (400) mg/vial = # of vials required
For a 60-kg patient: [(60 kg) × (10 mg/kg)] ÷ 300 (400) mg/vial = 2 vials of the 300-mg/vial solution or 1.5 vials of the 400-mg/vial solution. After patient is weighed and appropriate dose is calculated, remove sufficient vials from the refrigerator. Aseptic technique imperative. Reconstitute the 300-mg vial with 13 mL of SWFI and the 400-mg vial with 17 mL SWFI to obtain a solution with a final concentration of 25 mg/mL. Some back pressure may be experienced and is normal. To dissolve the lyophilized cake, hold vial upright and swirl the solution by rotating the vial. Gently invert the vial a few times to dissolve all the powder. Avoid vigorous agitation. Do Not Shake; should dissolve in less than 10 minutes. Allow the reconstituted solution to stand for 5 to 10 minutes. Solution should be colorless to slightly yellow. Do not use if opaque particles, discoloration, or other foreign particles are present. Vials contain overfill. Withdraw the calculated dose and further dilute in 230 mL of NS or D5W. The volume of NS or D5W can be adjusted so as not to exceed 5 mL/kg of patient weight at any given dose of elotuzumab. Gently invert the infusion bag to mix the solution. Infusion bags must be made of polyvinylchloride (PVC) or polyolefin.
Filters:
Must be administered through an infusion set with an in-line, sterile, nonpyrogenic, low–protein binding filter (pore size 0.2 to 1.2 micrometers).
Storage:
Refrigerate at 2° to 8° C (36° to 46° F) in original carton to protect from light until time of use. Do not freeze or shake. The diluted product may be stored for up to 24 hours if refrigerated and protected from light (a maximum of 8 of the 24 hours can be at RT and room light). Bring to room temperature before infusion and use immediately. Infusion should be completed within 24 hours of reconstitution. Discard any unused product remaining in vials.
Compatibility
Manufacturer states, “Do not mix elotuzumab with, or administer as an infusion with, other medicinal products.” Compatible only with IV bags/containers made of polyvinylchloride (PVC) or polyolefin.
Rate of administration
For IV infusion only. Use of an infusion pump recommended. Initiate the infusion rate at 0.5 mL/min. Increase in a stepwise fashion (see the following chart) if no infusion reactions develop. Interrupt elotuzumab for Grade 2 or higher infusion reactions.
| Recommended Infusion Rates for Elotuzumab | ||
| Cycle 1, Dose 1 | Cycle 1, Dose 2 | Cycle 1, Doses 3 and 4, and All Subsequent Cycles |
| Time Interval Rate | Time Interval Rate | Rate |
| 0 to 30 min: 0.5 mL/min | 0 to 30 min: 1 mL/min | 2 mL/min |
| 30 to 60 min: 1 mL/min | 30 min or more: 2 mL/min | 2 mL/min |
| 60 min or more: 2 mL/min | — | 2 mL/min |
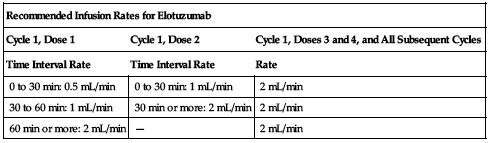
Adjust the infusion rate following a Grade 2 or higher infusion reaction; see Dose Adjustments. The maximum infusion rate should not exceed 2 mL/min; however, in patients who have received 4 cycles of elotuzumab, the infusion rate may be increased to a maximum of 5 mL/min.
Actions
Elotuzumab is a humanized recombinant IgG1 monoclonal antibody that specifically targets the SLAMF7 (signaling lymphocytic activation molecule family member 7) protein. SLAMF7 is expressed on myeloma cells independent of cytogenetic abnormalities. It is also expressed on natural killer cells, plasma cells, and some specific immune cell subsets of differentiated cells within the hematopoietic lineage. Elotuzumab targets SLAMF7 on myeloma cells and facilitates the interaction with natural killer cells to mediate the killing of myeloma cells through antibody-dependent cellular cytotoxicity (ADCC). In preclinical models, the combination of elotuzumab and lenalidomide resulted in enhanced activation of natural killer cells. When given in combination with lenalidomide/dexamethasone, approximately 97% of the maximum steady-state concentration is predicted to be eliminated with a geometric mean (CV%) of 82.4 days.
Indications and uses
Treatment of patients with multiple myeloma who have received one to three prior therapies. Used in combination with lenalidomide and dexamethasone.
Contraindications
Manufacturer states, “None.” Manufacturer recommends consulting the prescribing information for lenalidomide and dexamethasone before starting therapy.
Precautions
For IV infusion only. ■ Administered under the direction of a physician knowledgeable in its use in a facility with adequate diagnostic and treatment facilities to monitor the patient and respond to any medical emergency. ■ Can cause infusion reactions. Reports of infusion reactions during clinical trials were Grade 3 or lower, and most occurred during the first dose; see Monitor. ■ Infections, including opportunistic infections, have occurred; some resulted in discontinuation of therapy or fatalities. ■ Second primary malignancies, including hematologic malignancies, solid tumors, and skin cancers, have been reported. ■ Hepatotoxicity (elevations in liver enzymes [ALT, AST] greater than 3 times the ULN, total bilirubin greater than 2 times the ULN, and alkaline phosphatase less than 2 times the ULN) has been reported. ■ Can affect the determination of complete response and of disease progression in some patients with IgG kappa myeloma protein; see Drug/Lab Interactions. ■ Clinically significant differences in the pharmacokinetics of elotuzumab were not observed based on age, gender, race, baseline LDH, albumin, renal impairment ranging from mild to severe (including ESRD with or without dialysis), and mild hepatic impairment. Pharmacokinetics of elotuzumab in patients with moderate to severe hepatic impairment is unknown. ■ A therapeutic protein; has the potential for immunogenicity.
Monitor:
Premedication required; see Usual Dose. ■ Obtain baseline CBC and differential and monitor as indicated during therapy. ■ Obtain baseline liver enzymes (e.g., ALT, AST, bilirubin, and alkaline phosphatase) and monitor periodically. ■ Stop elotuzumab if Grade 3 or higher liver enzyme elevation occurs. Continuation of treatment may be considered after return to baseline values. ■ Monitor vital signs. ■ Monitor for S/S of infusion reactions; bradycardia, chills, fever, hypertension, and hypotension were most common. In patients who experience an infusion reaction, monitor vital signs every 30 minutes for 2 hours after the end of the elotuzumab infusion. ■ Immediately interrupt the infusion for infusion reactions of Grade 2 or higher; see Dose Adjustments and Antidote. ■ Monitor for S/S of infection. ■ Monitor for S/S of second primary malignancies.
Patient education:
Review manufacturer’s medication guide. ■ Because of the combination use with lenalidomide, pregnancy must be avoided. Effective contraception is required during treatment for men and women of reproductive potential. Consult with a health professional for specific information. ■ Premedication required to reduce the risk of infusion reactions. ■ Immediately report any S/S of infusion-related reactions (e.g., chills, difficulty breathing, fever, rash); may occur within 24 hours of an infusion. ■ Elotuzumab can affect the results of some tests, including testing for complete response, and additional testing may be indicated. ■ Second primary malignancies, including skin cancer, may occur; monitoring is required. Report unusual symptoms. ■ Report S/S of hepatotoxicity (abdominal pain, bruising, fatigue, itching, jaundice).
Maternal/child:
Combination drug regimen can cause fetal harm; avoid pregnancy; see Patient Education. ■ Discontinue breast-feeding. Safety for use during breast-feeding is unknown. Combination drug regimen may have serious effects in a breast-feeding infant. ■ Safety and effectiveness for use in pediatric patients not established.
Elderly:
No overall differences in safety or efficacy have been reported between elderly patients and younger adults.
Drug/lab interactions
No drug interaction studies have been performed. ■ Elotuzumab may be detected on both the serum protein electrophoreses (SPEP) and immunofixation (IFE) assays used for the clinical monitoring of endogenous M protein and can interfere with correct response classification. This interference can affect the determination of complete response and possibly cause a relapse from complete response in patients with IgG kappa myeloma protein.
Side effects
The most frequently reported side effects include constipation, cough, decreased appetite, diarrhea, fatigue, fever, infusion reactions, nasopharyngitis, peripheral neuropathy, pneumonia, and upper respiratory tract infections. Altered mood, bradycardia, cataracts, chest pain, headache, hepatotoxicity, hypersensitivity/infusion reactions, hypertension, hypoesthesia, hypotension, infections, night sweats, oropharyngeal pain, pain in extremities, second primary malignancies, tachycardia, weight loss, and vomiting have also been reported. Reported laboratory abnormalities included elevated alkaline phosphatase, hyperglycemia, hyperkalemia, hypoalbuminemia, hypocalcemia, leukopenia, lymphopenia, low bicarbonate, and thrombocytopenia.
Antidote
Notify physician of any side effects; most will be treated symptomatically. If a Grade 2 or higher infusion reaction occurs, interrupt elotuzumab infusion and institute appropriate medical and supportive measures. Severe infusion reactions may require permanent discontinuation of elotuzumab therapy. Stop elotuzumab upon Grade 3 or higher elevation of liver enzymes. After return to baseline values, continuation of therapy may be considered. Discontinue administration at the first sign of a serious hypersensitivity reaction and treat as indicated (e.g., oxygen, diphenhydramine, epinephrine, corticosteroids, vasopressors, and/or fluids). Resuscitate as necessary. Does not appear to be removed by dialysis.
Enalaprilat 
(en-AL-ah-prill-at)
ACE inhibitor
Antihypertensive
Vasodilator
pH 6.5 to 7.5
Usual dose
1.25 mg every 6 hours. Doses up to 5 mg every 6 hours have been tolerated for up to 36 hours, but clinical studies have not shown a need for dosage over 1.25 mg. Additional doses of 1.25 mg may be given every 6 hours except in dialysis patients. Dosage is the same when converting from oral to IV therapy. Resume oral therapy as soon as tolerated. See Precautions.
Pediatric dose
0.625 to 1.25 mg every 6 hours. See Maternal/Child.
Dose adjustments
Reduce initial dose to 0.625 mg in patients taking diuretics, patients with CHF, hyponatremia, severe volume or salt depletion, a CrCl less than 30 mL/min, and dialysis patients; see Rate of Administration. If the 0.625 dose is not clinically effective after 1 hour, it may be repeated. ■ Blood levels markedly increased in the elderly; dose selection should be cautious. Consider decreased cardiac, hepatic, and renal function; concomitant disease; or other drug therapy. ■ See Drug/Lab Interactions and Precautions.
Dilution
May be given undiluted through the port of a free-flowing infusion of NS, D5W, D5NS, D5LR, or Isolyte E. May also be diluted in up to 50 mL of any of the same solutions and given as an infusion.
Storage:
Store at CRT. Stable for up to 24 hours after dilution.
Compatibility (underline indicates conflicting compatibility information)
Consider any drug NOT listed as compatible to be INCOMPATIBLE until consulting a pharmacist; specific conditions may apply.
One source suggests the following compatibilities:
Additive:
Dextran 40, dobutamine, dopamine, heparin, hetastarch in NS (Hespan), meropenem (Merrem IV), nitroglycerin IV, nitroprusside sodium, potassium chloride (KCl).
Y-site:
Allopurinol (Aloprim), amifostine (Ethyol), amikacin, aminophylline, ampicillin, ampicillin/sulbactam (Unasyn), aztreonam (Azactam), bivalirudin (Angiomax), butorphanol (Stadol), calcium gluconate, cefazolin (Ancef), ceftaroline (Teflaro), ceftazidime (Fortaz), chloramphenicol (Chloromycetin), cisatracurium (Nimbex), cladribine (Leustatin), clindamycin (Cleocin), dexmedetomidine (Precedex), dextran 40, dobutamine, docetaxel (Taxotere), dopamine, doripenem (Doribax), doxorubicin liposomal (Doxil), erythromycin (Erythrocin), esmolol (Brevibloc), etoposide phosphate (Etopophos), famotidine (Pepcid IV), fenoldopam (Corlopam), fentanyl, filgrastim (Neupogen), ganciclovir (Cytovene IV), gemcitabine (Gemzar), gentamicin, granisetron (Kytril), heparin, hetastarch in electrolytes (Hextend), hetastarch in NS (Hespan), hydrocortisone sodium succinate (Solu-Cortef), labetalol, lidocaine, linezolid (Zyvox), magnesium sulfate, melphalan (Alkeran), meropenem (Merrem IV), methylprednisolone (Solu-Medrol), metronidazole (Flagyl IV), morphine, nafcillin (Nallpen), nicardipine (Cardene IV), nitroprusside sodium, oxaliplatin (Eloxatin), pemetrexed (Alimta), penicillin G potassium, phenobarbital (Luminal), piperacillin/tazobactam (Zosyn), potassium chloride (KCl), potassium phosphates, propofol (Diprivan), ranitidine (Zantac), remifentanil (Ultiva), sodium acetate, sulfamethoxazole/trimethoprim, teniposide (Vumon), thiotepa, tobramycin, vancomycin, vinorelbine (Navelbine).
Rate of administration
A single dose must be evenly distributed over 5 minutes. Extend rate of infusion up to 1 hour in patients at risk for severe hypotension (e.g., heart failure, hyponatremia, high-dose diuretic therapy, recent intensive diuresis or increase in diuretic dose, renal dialysis, or severe volume and/or salt depletion of any etiology).
Actions
An antihypertensive agent. An angiotensin-converting enzyme inhibitor that prevents conversion of angiotensin I to angiotensin II. Peripheral arterial resistance is reduced in hypertensive patients. In patients with heart failure, significant reduction in pulmonary capillary wedge pressure (preload), peripheral vascular resistance (afterload), BP, and heart size occurs, as well as an increase in cardiac output (stroke index) and exercise tolerance time. Initial response may take 15 minutes to 1 hour. Peak BP reduction occurs in 1 to 4 hours, and effects last up to 6 hours. Peak effects of subsequent doses may be greater than the initial dose. Excreted in urine. Crosses placental barrier. Secreted in breast milk.
Indications and uses
Treatment of hypertension when oral therapy is not practical. ■ Heart failure not adequately responsive to diuretics and digoxin. Enalaprilat is used in addition to digoxin and diuretics. ■ Hypertensive emergencies (effects are variable).
Unlabeled uses:
Treatment of hypertension or renal crisis in scleroderma.
Contraindications
Hypersensitivity to enalaprilat or its components, a history of angioedema related to previous treatment with an ACE inhibitor, or hereditary or idiopathic angioedema.
Precautions
Has been used IV for up to 7 days. ■ Use caution in patients with a history of angioedema (see Contraindications), aortic stenosis, or hypertrophic cardiomyopathy. ■ Use caution in patients with collagen vascular disease and renal disease; neutropenia and/or agranulocytosis have been reported. Monitoring of WBC may be indicated. ■ Use caution in surgery, with anesthesia, or with agents that produce hypotension. ■ May rarely cause a syndrome that starts with cholestatic jaundice, progresses to hepatic necrosis, and may progress to death. Discontinue in patients who develop elevated liver enzymes or jaundice. ■ ACE inhibitors often cause a persistent, nonproductive cough, which should resolve when drug is discontinued. ■ Average dose for conversion to oral therapy is 5 mg/day as a single dose. When a reduced dose of enalaprilat IV has been indicated (e.g., diuretics, impaired renal function, dialysis), reduce initial oral dose to 2.5 mg/day as a single dose. Adjust either by patient response. ■ Patients sensitive to one ACE inhibitor may be sensitive to another. ■ See Monitor and Drug/Lab Interactions.
Monitor:
Monitor vital signs very frequently. May cause precipitous drop in BP following the first dose. ■ Use extreme caution in fluid-depleted patients. Patients with congestive heart failure may become hypotensive at any time. Arrhythmias or conduction defects may occur. ■ Monitor BUN and SCr. An increase in either may require a decrease in dose of enalaprilat or discontinuation of a diuretic. ■ Diuretics given concomitantly may cause a precipitous drop in BP within the first hour of the initial dose; observe the patient closely. Severe dietary salt restriction or dialysis will aggravate this effect. ■ May cause oliguria or progressive azotemia in patients with severe congestive heart failure whose renal function is dependent on the activity of the renin-angiotensin-aldosterone system. Acute renal failure and death are possible. ■ May cause hyperkalemia. May cause a significant increase in serum potassium with potassium-sparing diuretics or potassium supplements. Use with caution and only in documented hypokalemia. Use salt substitutes with caution. Monitor serum potassium levels. ■ Monitoring of WBC may be indicated in patients with collagen vascular disease or renal disease. ■ See Drug/Lab Interactions.
Patient education:
Consider birth control options. ■ May cause dizziness; avoid sudden changes in posture and request assistance for ambulation if necessary.
Maternal/child:
Avoid pregnancy; Category C (first trimester) and Category D (second and third trimester). Can cause fetal and neonatal morbidity and death. Infants exposed to ACE inhibitors during the first trimester of pregnancy may have an increased risk of major congenital malformations. If pregnancy occurs, discontinue immediately; many alternate antihypertensive agents. ■ Observe any infant with in utero exposure for hypotension, oliguria, and hyperkalemia. ■ Has caused reversible acute renal failure in a premature infant whose mother received enalaprilat. ■ Safety for use in breast-feeding not established. ■ Safety for use in pediatric patients not established but has been used. ■ May contain benzyl alcohol, which has been associated with a fatal “gasping syndrome” in neonates.
Elderly:
Dose selection should be cautious; see Dose Adjustments and Precautions/Monitor. ■ May be less sensitive to effects due to a decrease in plasma renin activity or more sensitive to hypotensive effects due to increased blood levels (decreased renal excretion).
Drug/lab interactions
Use caution in surgery, with anesthesia, or with any agents that produce hypotension. ■ May be used concomitantly with other antihypertensive agents (e.g., thiazide diuretics [chlorothiazide (Diuril)]). Effects are additive. ■ Diuretics given concomitantly may cause a precipitous drop in BP. ■ May cause hyperkalemia with potassium-sparing diuretics (e.g., spironolactone [Aldactone], triamterene [Dyrenium], amiloride [Midamor]), potassium supplements, potassium-containing salt substitutes, or low-salt milk. ■ Use caution and consider lower doses when administering nitroglycerin, nitroprusside sodium, other nitrates, or other vasodilators (e.g., hydralazine). ■ In patients with compromised renal function, concurrent use with NSAIDs (e.g., ibuprofen [Advil, Motrin], naproxen [Aleve, Naprosyn]) may result in further deterioration of renal function. ■ Concurrent use with NSAIDs may also decrease the hypotensive effects of enalaprilat by inhibiting the renal prostaglandin synthesis and/or by causing sodium and fluid retention. ■ May increase lithium concentration, resulting in lithium toxicity. ■ Interaction with some imaging agents (e.g., iodohippurate, technetium) may render diagnostic renal function tests inconclusive. ■ May decrease hemoglobin and hematocrit slightly. ■ See Precautions and Monitor.
Side effects
Abdominal pain, angioedema, anosmia (absence of sense of smell), atrial fibrillation, bradycardia, chest pain, conjunctivitis, cough (persistent, dry), diarrhea, dizziness, dry eyes, dyspnea, eosinophilic pneumonitis, fatigue, flank pain, gynecomastia, headache, hepatotoxicity, herpes zoster, hoarseness, hyperkalemia, hypotension (severe), impotence, insomnia, muscle cramps, nausea, palpitations, paresthesias, photosensitivity, pneumonia, pruritus, pulmonary edema, pulmonary embolism and infarction, pulmonary infiltrates, rash, Raynaud’s phenomenon, renal failure (reversible), rhinorrhea, somnolence, sore throat, taste disturbances, tearing, toxic epidermal necrolysis, vomiting. Anaphylaxis has been reported.
Antidote
For minor side effects, notify the physician. Most will be tolerated or treated symptomatically. If symptoms progress or any major side effect occurs (angioedema, precipitous hypotension, hyperkalemia), discontinue drug and notify the physician immediately. Hypotension should respond to IV fluids if the patient’s condition allows their use. Other drugs in the regimen may need to be discontinued or the dosage reduced. Epinephrine, diphenhydramine (Benadryl), and hydrocortisone may be used to treat angioedema. Maintain the patient as indicated. If cardiac arrhythmias occur, treat appropriately. Hemodialysis may be useful in toxicity.
Epinephrine hydrochloride
(ep-ih-NEF-rin hy-droh-KLOR-eyed)
Adrenalin Chloride
Cardiac stimulant
Bronchodilator
Antiallergic
Vasopressor
pH 2.5 to 5
Usual dose
Hypersensitivity reactions or bronchospasm:
0.1 to 0.25 mg (1 to 2.5 mL of a 0.1 mg/mL concentration). Start with a small dose, giving only as much as required to alleviate undesirable symptoms, and repeat as necessary (usually every 20 to 30 minutes), gradually increasing dose depending on need. Another source suggests 0.2 to 0.5 mg of 0.1 mg/mL concentration. May be repeated as necessary.
Bradycardia:
AHA guidelines recommend epinephrine infusion 2 to 10 mcg/min; titrate to desired effect. Indicated if atropine is ineffective. Alternately, transcutaneous pacing or dopamine infusion 2 to 10 mcg/kg/min may be used.
Cardiac arrest:
AHA guidelines recommend 1 mg (10 mL of a 0.1 mg/mL concentration) IV; may repeat every 3 to 5 minutes. Follow each dose with a 20-mL IV flush to ensure delivery to systemic circulation. See Compatibility. Doses up to 0.2 mg/kg have been used for specific indications (beta-blocker or calcium channel blocker overdose). May also be given as a continuous infusion by adding 1 mg of epinephrine (1 mL of a 1 mg/mL solution) to 500 mL NS or D5W. Begin with an infusion rate of 0.1 to 0.5 mcg/kg/min and titrate to response. The dose for a 70-kg patient would be 7 to 35 mcg/min. Higher doses of epinephrine are controversial.
Endotracheal:
A diluted solution may be given through the endotracheal tube before an IV is established. AHA guidelines recommend 2 to 2.5 mg (of a 1 mg/mL solution) diluted in 10 mL NS. Another source suggests administering the IV dose through the endotracheal tube if an IV line has not been established.
Vasopressor or maintenance dose:
1 to 10 mcg/min titrated to desired response. AHA guidelines recommend that epinephrine be used to treat symptomatic bradycardia after atropine as an alternative infusion to dopamine or to treat severe hypotension when atropine and transcutaneous pacing fail, when hypotension accompanies bradycardia, or with a phosphodiesterase enzyme inhibitor. For profound bradycardia or hypotension, 2 to 10 mcg/min may be given as an infusion (1 mg of 1 mg/mL concentration in 500 mL NS or D5W) at a rate of 0.1 to 0.5 mcg/kg/min titrated to response.
Pediatric dose
See Maternal/Child.
Hypersensitivity reactions or bronchospasm in infants and children:
0.01 mg/kg (0.1 mL/kg of a 0.1 mg/mL concentration). May repeat at 20-minute to 4-hour intervals. One source suggests a maximum dose of 0.3 mg, another 0.5 mg. Usually given SC as a 1 mg/mL concentration.
Severe anaphylactic shock in infants and children:
One source suggests 0.1 mg IV of a 0.01 mg/mL concentration (0.1 mL of a 1 mg/mL concentration diluted in 10 mL NS) given over 5 to 10 minutes. Another source suggests 0.01 mL/kg of a 1 mg/mL concentration SC. Maximum 0.3 mL/dose. Repeat every 15 minutes as needed.
Bradycardia in infants and children:
AHA guidelines recommend 0.01 mg/kg (0.1 mL/kg of a 0.1 mg/mL concentration) to treat symptomatic bradycardia. If IV access is not readily available, AHA guidelines recommend 0.1 mg/kg (0.1 mL/kg) of a 1 mg/mL concentration via ET.
Asystolic or pulseless arrest in infants and children:
AHA guidelines recommend 0.01 mg/kg (0.1 mL/kg of a 0.1 mg/mL concentration). Another source recommends 0.01 mg/kg of a 0.1 mg/mL concentration and suggests that the first dose should not exceed 1 mg (10 mL of a 0.1 mg/mL concentration). Repeat every 3 to 5 minutes during arrest. Up to 0.1 to 0.2 mg/kg may be used if initial doses are ineffective. May be given via ET (0.1 mg/kg [0.1 mL/kg of a 1 mg/mL concentration]) every 3 to 5 minutes until IV established, then begin with first IV dose. A third source suggests 0.01 to 0.03 mg/kg (0.1 to 0.3 mL/kg) of a 0.1 mg/mL concentration initially. May repeat every 3 to 5 minutes in neonates. In infants and children subsequent doses of 0.1 mg/kg every 3 to 5 minutes may be given if needed. Prepare an infusion and titrate from 0.1 to 1 mcg/kg/min to desired effect. Use upper dosing range if asystole is present. With higher dose, be aware of preservative content to avoid toxicity.
Dose adjustments
See Drug/Lab Interactions. ■ Doses larger than 1 mg may not be indicated in patients over 65 years of age and patients in ventricular fibrillation.
Dilution
New changes in labeling eliminate the use of ratios; what was previously a 1:1,000 solution will now be referred to only as a 1 mg/mL solution, a 1:10,000 solution will now be referred to only as a 0.1 mg/mL solution, and a 1:100,000 solution will now be referred to only as a 0.01 mg/mL solution.
Check label. Not all epinephrine solutions can be given IV. The 1 mg/mL strength is for SC or IM use only. It must be further diluted with at least 10 mL of NS to prepare a 0.1 mg/mL solution before IV use.
IV injection:
Available prediluted (0.1 mg/mL) in 10-mL syringes. Available in a 30-mL vial (30 mg [1 mg/mL solution]) to facilitate larger doses or continuous infusion. Each 1 mg (1 mL) of 1 mg/mL solution must be diluted in at least 10 mL of NS to prepare a 0.1 mg/mL solution.
Infusion:
For occasional use as a vasopressor or for maintenance, epinephrine may be further diluted in 250 to 500 mL D5W; see the following chart. Give through Y-tube or three-way stopcock of infusion set. See chart on inside back cover for additional compatible solutions.
| Epinephrine HCl Infusion Rates | ||||||
| Desired Dose | 1 mg in 500 mL D5W (2 mcg/mL) | 1 mg in 250 mL D5W 2 mg in 500 mL D5W (4 mcg/mL) | ||||
| mcg/min | mcg/hr | mL/min | mL/hr | mcg/hr | mL/min | mL/hr |
| 1 | 60 | 0.5 | 30 | 60 | 0.25 | 15 |
| 2 | 120 | 1 | 60 | 120 | 0.5 | 30 |
| 3 | 180 | 1.5 | 90 | 180 | 0.75 | 45 |
| 4 | 240 | 2 | 120 | 240 | 1 | 60 |
| 5 | 300 | 2.5 | 150 | 300 | 1.25 | 75 |
| 6 | 360 | 3 | 180 | 360 | 1.5 | 90 |
| 7 | 420 | 3.5 | 210 | 420 | 1.75 | 105 |
| 8 | 480 | 4 | 240 | 480 | 2 | 120 |
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


