Chapter 4 Disorders of the Endocrine System
When you have completed this chapter you will be able to
THE ENDOCRINE SYSTEM
The endocrine system is made up of endocrine glands (and some endocrine tissue), which are widely dispersed throughout the body1. The major endocrine glands are the hypothalamus, the pituitary gland, the pineal gland, the thyroid gland, the parathyroid glands, the thymus gland, the pancreatic islets, the adrenal glands, and the ovaries and testes1,2. Unlike the exocrine glands that transport their non-hormonal chemical secretions within ducts and ultimately to the exterior of the body, the endocrine glands are ductless glands that secrete hormones directly into tissue fluid. Some hormones are secreted into the bloodstream and transported to distant sites where they exert their action1. Other hormones not secreted into the bloodstream act locally. Each hormone binds to specific hormone receptors located on target cells throughout the body. Hormones may have paracrine or autocrine action; in paracrine signalling, the hormone acts on nearby target cells and in autocrine signalling, the hormone acts on the same cells that produce it3.
Hormones are either amino acid-based or steroid-based1. Most hormones are of the amino acid type1; they are water soluble with relatively short half-lives4 and are carried to the target cell dissolved in plasma. Steroid hormones are derived from cholesterol1, they are insoluble in plasma and are transported to their target cell bound to a carrier protein, resulting in a longer half-life3,4.
Hormones exert their effect by binding to receptor sites, which are located on the inside or outside of the target cells3. Steroid hormones bind to specific receptors inside target cells. This interaction can stimulate or inhibit genetic transcription, which modifies protein production in the target cell4,5. Water-soluble hormones bind to protein receptors located on the outside of the target cell membrane, activating distinctive types of cellular molecules called second messengers. Second messengers trigger a series of molecular reactions, which alter the physiological state of the cell5. By either method of binding, hormones act as chemical messengers to influence the metabolic activity of the target cell3,4.
Hormone production by each endocrine gland is finely balanced. Endocrine disorders are usually manifested by an overproduction or an underproduction of a specific hormone, which alters the function of various body systems. Hormones have multiple and wide ranging influences on body processes including reproduction, growth and development, stress responses, maintenance of fluid and electrolyte balance and nutrients in the blood, and regulation of metabolism1. Some common endocrine disorders are discussed below.
DIABETES
The most obvious sign of diabetes is excessive urination or polyuria. Diabetes was given its name by the Greeks after their word for siphon or ‘to flow through’. There are two distinct types of diabetes: diabetes insipidus and diabetes mellitus. The word insipidus is derived from the Latin word meaning ‘having no flavour’ whereas mellitus means ‘sweetened or honey-like’ to indicate the urine was sweet. Although these two types of diabetes are distinctly different, they have a number of similarities. Hormonal deficiencies or hormonal resistance causes both types of diabetes and common symptoms include excessive thirst and frequent urination6. Diabetes mellitus is a relatively common condition, whereas diabetes insipidus is relatively rare7.
DIABETES INSIPIDUS
There are two main types of diabetes insipidus (DI): central (neurogenic) and nephrogenic. In central DI, there is a deficiency of antidiuretic hormone (ADH), also called vasopressin8. The deficiency of ADH production by the ADH secretory neurones in the hypothalamus may be a primary disorder (e.g., congenital), or a secondary disorder (e.g., head trauma, tumours, inflammation or haemorrhage)9. In nephrogenic diabetes insipidus there is normal production of ADH. However, the kidney’s ability to respond to ADH is impaired. This may also be a primary disorder (e.g., genetic disorder), or secondary to renal disease, hypokalaemia, hypercalcaemia or to drugs that inhibit the peripheral action of ADH, such as lithium9.
Mr Smolcic has nephrogenic DI related to lithium. Lithium is frequently prescribed for the treatment of psychiatric conditions, and may result in lithium-induced DI, which is the most common cause of DI. In view of the hypernatraemia (high serum sodium), Mr Smolcic was commenced on a dietary restriction of sodium. He was given 4 mcg of desmopressin acetate intravenously, which was associated with a modest decrease in serum sodium and in urine output. For long-term management, Mr Smolcic was prescribed a daily dose of amiloride HCl 5mg, a daily dose of hydrochlorothiazide 50mg, and indomethacin 25mg three times daily9.
PATHOPHYSIOLOGY
Diabetes insipidus is caused by ineffective or deficient secretion of ADH10. Normally ADH increases solute-free water reabsorption through the tubular cells of the kidneys. The water is returned to the circulation, leading to a decrease in plasma osmolality, an increase in urine osmolality and low urine volumes. Individuals who are deficient in ADH (central DI) and those whose renal tubules are insensitive or resistant to the effects of ADH (nephrogenic DI) are compromised in their ability to conserve solute-free water8–10.
CLINICAL MANIFESTATIONS
The clinical manifestations of DI include an excessive and dilute urine output, which may range from 3 to 18 litres per day7–9. Other clinical features include polydipsia (excessive thirst), which is an attempt to compensate for the excessive water loss. Nocturia is a common symptom, which can lead to sleep deprivation and fatigue7–9. If the patient does not have an adequate fluid intake, dehydration and hypernatraemia may ensue.
DIAGNOSTIC TESTS
DI may be diagnosed by a urine specific gravity of less than 1.005 and a urine osmolality of less than 200mOsm/kg7. Serum osmolality may be within the normal range (280 to 295mOsm/kg). There will, however, be a noticeable rise in serum osmolality if water is not readily available or if the patient’s thirst mechanisms are inadequate. A water deprivation test may be used in the diagnosis of DI, and may also be used to differentiate between central and nephrogenic forms of DI9,11. Careful monitoring is necessary during this diagnostic procedure due to the risk of severe dehydration. The test involves withholding fluid intake and testing urine and plasma osmolality10. This is followed by the administration of desmopressin and further testing of urine and plasma osmolality. In the patient with central DI, urine osmolality is lower than plasma osmolality. However after the administration of desmopressin, urine osmolality increases dramatically10. In nephrogenic DI, urine osmolality is also less than plasma osmolality, with only a slight increase in urine osmolality after the administration of desmopressin12.
TREATMENT AND APPLIED PHARMACOLOGY
The treatment of the patient with DI requires fluid replacement as well as correction of hypernatraemia and other electrolyte abnormalities. For patients with central DI, the treatment of choice is desmopressin, a synthetic analogue of ADH13. In nephrogenic DI, the effectiveness of desmopressin is usually limited14, as the underlying problem is not ADH deficiency but an impaired renal response to ADH. Both central and nephrogenic DI will partially respond to the use of thiazide diuretics, such as hydrochlorothiazide or amiloride. These drugs act by depleting total body sodium and thus increasing the isotonic absorption of water in the proximal tubule of nephrons7. In addition to the promotion of sodium loss by the use of thiazide diuretics, dietary sodium restriction is also recommended for the correction of hypernatraemia. Non-steroidal anti-inflammatory drugs (NSAIDs) such as indomethacin are sometimes used in the treatment of nephrogenic DI during the acute phase15; these act through inhibiting prostaglandin synthesis, which regulates glomerular blood flow and reduces urine output12.
DIABETES MELLITUS
Diabetes mellitus (DM) is broadly described as a chronic, metabolic disorder characterised by abnormalities in carbohydrate, protein and fat metabolism resulting from defects in insulin secretion, insulin action, or both16. The three most common forms of DM are type 1, type 2 and gestational diabetes. The classification of type 1 DM as juvenile onset and type 2 DM as mature onset is not an accurate reflection of the epidemiology of this group of disorders. Hence, the use of these terms or ‘insulin-dependent diabetes mellitus’ and ‘non-insulin-dependent diabetes mellitus’ and their acronyms ‘IDDM’ and ‘NIDDM’ is obsolete as these terms can be inaccurate and misleading16,17.
Diabetes mellitus is a serious and growing global public health issue contributing to significant premature mortality, morbidity, disability and loss of potential years of life18. More than 7% of the Australian adult population have DM. However, the prevalence increases to 23% in those aged 75 years or older and is estimated to be 10 to 30% among the indigenous communities and people from the Pacific Islands and some Asian countries19. In New Zealand, the rate of DM among adults of European descent is 3.1% but more than 8% among those of MaF8ori and Pacific Island descent20. Australia experiences very high rates of type 1 DM compared with most countries of the world, and it is ranked as one of the most common serious childhood diseases21. Type 2 DM is the major form of diabetes in Australia and is a common disorder among people over the age of 40 years19. With the increase in paediatric obesity, Type 2 DM is also emerging to be a health issue among children and adolescents.
Type 1 DM is primarily caused by immune-mediated destruction of beta cells of the pancreas, resulting in minimal or no insulin production22. Hence the person with type 1 DM requires life-long treatment with insulin. Type 2 DM results from a combination of insulin resistance and a defect in insulin secretion; the person with type 2 DM may not require insulin administration. There is a strong association between type 2 DM and excess body fat, with an estimated 80–90% of people with type 2 DM being overweight or obese23. It has been suggested that the problems of obesity and type 2 DM, share some of the same causative factors, for example, excessive energy intake and low physical activity24. There is evidence that type 2 DM can be prevented in high-risk individuals by lifestyle interventions, such as weight loss through diet and exercise25,26.
Gestational diabetes mellitus (GDM) is defined as any degree of glucose intolerance during pregnancy16. Gestational diabetes is similar to type 2 DM in that it also occurs at higher rates in some populations, including the indigenous populations of Australasia, America, Asia and the Pacific Islands.Although glucose tolerance usually returns to normal shortly after delivery, up to 50% of those who develop gestational diabetes will develop type 2 DM within ten years27.
Case study – type 1 diabetes mellitus
Penny has type 1 DM and is experiencing an acute complication known as diabetic ketoacidosis (DKA). She was commenced on an intravenous infusion of normal saline, followed by an insulin infusion. Regular monitoring of serum glucose, urea, creatinine, electrolytes and ABGs was undertaken. Once stabilised, Penny was transferred to the high dependency unit for further management. Both DKA and hyperosmolar hyperglycaemic state (HHS) are hyperglycaemic emergencies that are acute complications of diabetes mellitus28. The magnitude of metabolic acidosis is usually more pronounced in DKA whereas the magnitude of dehydration is usually more severe in HHS. DKA is most often seen with type 1 DM, whereas HHS is more likely to occur with type 2 DM. The most common precipitating factor in both DKA and HHS is infection29. Other precipitating factors include pancreatitis, cardiovascular events, trauma and drugs. DKA has a mortality rate of 5% compared to HHS, which has a mortality rate of 15%29.
PATHOPHYSIOLOGY OF DKA AND HHS
The pathophysiological basis of DKA and HHS is shown in Figure 4.1. The underlying pathophysiological mechanism in both DKA and HHS is a deficiency in insulin resulting in an increase in the release of counter-regulatory hormones (glucagon, catecholamines, cortisol and growth hormone)28,30. Insulin deficiency and increased counter-regulatory hormones cause hyperglycaemia by accelerating gluconeogenesis (production of glucose), glycogenolysis (breakdown of glycogen) and through decreased peripheral glucose use. As blood glucose levels increase there is a corresponding rise in serum osmolality, which creates an osmotic gradient, causing fluid to shift from the intracellular fluid compartment to the intravascular fluid compartment resulting in intracellular dehydration28,30.
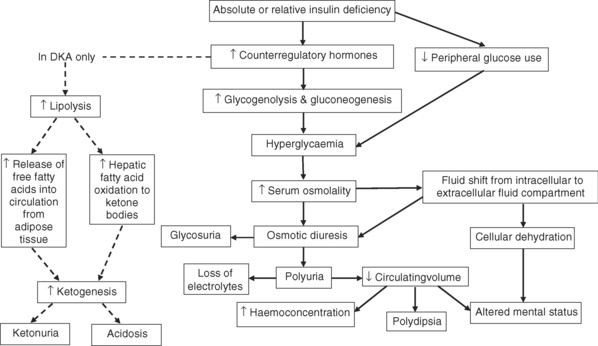
In DKA, the counter-regulatory hormones activate lipolysis (fat breakdown). Hormone-sensitive lipase causes the breakdown of triglycerides releasing free fatty acids. The liver takes up the free fatty acids and converts them to ketone bodies, a process predominantly stimulated by glucagon28. Ketone bodies are buffered by bicarbonate. However, the production of ketone bodies is in excess to bicarbonate levels resulting in ketosis, which is a state of metabolic acidosis28.
Ketosis is not a hallmark of HHS. The reason for the absence of ketosis remains unclear, although factors including lower levels of free fatty acids and higher levels of endogenous insulin reserve are two proposed explanations28.
CLINICAL MANIFESTATIONS
The clinical manifestations of both DKA and HHS include glycosuria, polyuria and altered mental status29. As blood glucose increases, the amount being filtered by the glomeruli in the kidneys exceeds the capacity of the renal tubule to reabsorb glucose. The consequence is urinary glucose loss, known as glycosuria. The high glucose content also exerts an abnormally high osmotic pressure within the renal filtrate, causing an osmotic diuresis, which induces excessive water and electrolyte loss. Dehydration, both intracellular and intravascular, occurs as a result of fluid shifts from the intracellular compartment, and hyperglycaemia-induced osmotic diuresis. Dehydration results in an increase in thirst leading to polydipsia (excessive water intake). However, this may be absent in elderly patients with HHS. Other presenting features of severe dehydration include dry mucous membranes, sunken eyeballs and poor skin turgor. Dehydration also increases haemoconcentration, which may lead to embolic complications such as deep vein thrombosis (DVT) and pulmonary embolism31. Neurological manifestations of dehydration include lethargy, stupor, confusion, aggression and coma. An altered mental status is more likely to occur in HHS because of the magnitude of hyperglycaemia-induced osmotic fluid loss.
In DKA only, the presence of ketone bodies results in the excretion of ketoacids in the urine (ketonuria). The presence of metabolic acidosis in DKA induces hyperventilation through the stimulation of central as well as peripheral chemoreceptors, which decreases the partial pressure of carbon dioxide in plasma. This type of laboured respiration is known as Kussmaul respiration. Abdominal pain and vomiting are frequently reported in patients with DKA but these are not typical manifestations of HHS29.
DIAGNOSTIC TESTS
Both clinical manifestations and laboratory tests are necessary to confirm the diagnosis of DKA and HHS. In DKA, plasma glucose levels are usually 14 mmol/L or greater30. The arterial blood pH is usually less than 7.3 and the serum bicarbonate is usually 15 mmol/L or less30. There is also evidence of ketonuria in DKA.
In HHS, hyperglycaemia is usually more severe than in DKA, with a plasma glucose level of 34 mmol/L or greater28. The profound hyperglycaemia that is experienced in HHS induces an hyperosmolar syndrome, with a serum osmolality of at least 320 mmol/kg29. The arterial blood pH is usually greater than 7.3 and there are either no ketones or only trace amounts in the urine of patients with HHS28.
The glutamic acid decarboxylase (GAD) antibodies test and the C-peptide test are sometimes used to assist in the differential diagnosis of type 1 and type 2 DM. The GAD antibodies test detects antibodies to insulin-producing beta cells16. These autoantibodies are elevated in people with type 1 DM, and are evidence of an autoimmune disorder. The C-peptide test is a blood test for a hormone produced in conjunction with endogenous insulin32. People with type 1 DM have low to unmeasurable levels of C-peptide, whereas in people with type 2 DM the levels are normal33.
TREATMENT AND APPLIED PHARMACOLOGY
The treatment of acute hyperglycaemic crisis aims to restore perfusion and improve circulatory volume, decrease serum glucose, correct acidosis, reverse lipolysis and proteolysis, correct electrolyte deficits, and avoid treatment complications including hypoglycaemia, cerebral oedema and hypokalaemia28–30. As a result of osmotic diuresis, patients with DKA and HHS usually present to hospital in a dehydrated state. Intravenous fluid therapy is necessary to restore both intravascular and intracellular volume. The initial fluid therapy is usually an infusion of normal saline. Subsequent fluid replacement depends on the state of hydration, serum electrolyte and glucose levels, and urinary output. In addition to correcting dehydration, fluid therapy will also improve hyperglycaemia, hyperosmolality and acidosis28–30.
The administration of insulin therapy is usually by means of continuous intravenous infusion via an automated infusion pump. Insulin therapy facilitates intracellular transport of glucose and the movement of water and electrolytes, such as potassium, magnesium and phosphate into the cell. It is important to aim for a gradual and steady fall in plasma glucose levels, as this decreases the risk of hypoglycaemia, hypokalaemia and cerebral oedema. In DKA, hyperglycaemia is usually corrected before ketoacidosis, therefore it is important to continue with insulin therapy to reverse and inhibit ketogenesis. A glucose infusion is commonly administered in conjunction with insulin therapy to avoid the complication of hypoglycaemia30.
During therapy for DKA or HHS, capillary blood glucose levels are measured every one to two hours at the bedside. Blood is also sampled regularly for laboratory measurements of serum electrolytes, glucose, urea and creatinine29,30. Regular monitoring of blood pH and serum bicarbonate is also recommended for patients with DKA29,30.
NURSING IMPLICATIONS
The implications of caring for a patient with acute hyperglycaemic crisis involve monitoring
Case study – type 2 diabetes mellitus
During hospitalisation, Mr Dass was prescribed routine medications for management of his MI (see Chapter 5). He was also commenced on subcutaneous insulin injections.
The MI experienced by Mr Dass is one of the major complications of type 2 DM. The most common cause of death in people with DM is from macrovascular disease, with coronary artery disease (CAD) the most prevalent34. The cardiovascular risk factors that are more common among people with type 2 DM include dyslipidaemia and hypertension34.
PATHOPHYSIOLOGY OF CHRONIC COMPLICATIONS OF DM
Diabetes mellitus is characterised by a number of chronic complications associated with metabolic dysfunction35. These complications can be broadly classified as macrovascular and microvascular complications. Macrovascular complications develop as a consequence of atherosclerotic disease of the large vessels, including cardiac, cerebral and peripheral vascular disease36. Microvascular complications usually only become evident in people with overt diabetes mellitus35. Examples of microvascular complications include diabetic retinopathy, nephropathy and neuropathy.
Macrovascular disease is usually evident during the pre-diabetic stage, as insulin resistance develops – a disorder associated with excess weight, ageing, infection and pregnancy37,38. Insulin resistance is the pre-diabetic stage of impaired glucose tolerance39 where there is decreased sensitivity of insulin receptors in the liver, adipose tissue and skeletal muscles. Patients with insulin resistance become hyperinsulinaemic to overcome the underlying defect of insulin resistance34. The association between insulin resistance, hyperinsulinaemia, hypertension, obesity, dyslipidaemia, the procoagulant state and cardiovascular disease is now well recognised34.
There is no single pathophysiological mechanism in the development of the chronic complications of DM. The postulated pathophysiological processes can be categorised as vascular abnormalities (e.g., endothelial dysfunctions, glucose-induced abnormalities, excessive glycation of circulating and membrane-bound proteins), and other mechanisms (e.g., abnormalities in growth factors and platelet dysfunction)34,36,40.
There is good evidence that the pathogenesis of the atherogenic lesion in macrovascular disease is endothelial cell dysfunction41. The causes of endothelial abnormalities include insulin resistance and hyperglycaemia41. High blood glucose levels inhibit the production of vasodilatory nitric oxide, which contributes to the procoagulant state in the blood34,41. A deficiency in nitric oxide promotes platelet aggregation, proliferation of smooth muscle and an increase in monocyte adhesiveness34,41. These alterations accelerate the formation of atherosclerosis and alter the structure and function of blood vessels.
Microvascular disease involves structural changes in the capillaries and small vessels and most commonly affects the retina causing diabetic retinopathy, the kidneys causing diabetic nephropathy, and the peripheral nerves causing diabetic neuropathy36. Risk factors for microvascular complications include poor glycaemic control, duration of diabetes, hypertension and microalbuminuria41. The pathogenesis of microvascular complications is multifactorial with metabolic and vascular involvement. There is evidence that glycation of body proteins results in the release of cytokines and other inflammatory mediators41. In neuropathy, the diminished blood supply to nerves results in axonal and myelin sheath damage. Oxidative stress, as a result of an increase in substances such as reactive oxygen species (ROS), is another pathogenic mechanism of diabetic complications42. ROS oxidise and damage deoxyribose nucleic acid (DNA), proteins and lipids resulting in tissue damage42.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


