Chapter 12 Development and transmission of HIV drug resistance
Introduction
HIV-1 drug resistance has emerged as a major problem that limits the effectiveness of antiviral drugs in treatment regimens. Many studies have shown that the development and transmission of drug-resistant HIV-1 is largely a consequence of incompletely suppressive antiretroviral regimens; HIV-1 drug resistance can significantly diminish the effectiveness and duration of benefit associated with combination therapy for the treatment of HIV/AIDS [1–6]. Resistance-conferring mutations in each of the HIV-1 reverse transcriptase (RT), protease (PR), and integrase (IN) genes may precede the initiation of therapy due to each of spontaneous mutagenesis and the spread of resistant viruses by sexual and other means. However, it is also generally believed that multiple drug mutations to any single or combination of antiretroviral agents (ARVs) are required in order to produce clinical resistance to most ARVs and that these are in fact selected following residual viral replication in the presence of incompletely suppressive drug regimens [7–9].
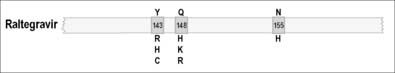
In the case of the protease inhibitors (PIs) [10–12], and most nucleoside analog reverse transcriptase inhibitors (NRTIs), the development of progressive high-level phenotypic drug resistance follows the accumulation of primary resistance-conferring mutations in each of the HIV-1 PR and RT genes [13–15]. Non-nucleoside reverse transcriptase inhibitors (NNRTIs) have low genetic barriers for the development of drug resistance and, frequently, a single primary drug resistance mutation to any one NNRTI may be sufficient to confer high-level phenotypic drug resistance to this entire class of ARVs [16, 17]. Raltegravir is the only currently approved integrase strand transfer inhibitor (INSTI) and has an intermediate genetic barrier for resistance, since an accumulation of two or more mutations in IN are generally required in order for this compound to lose its antiviral efficacy [18, 19].
Furthermore, differences have also been reported in regard to the development and evolution of antiretroviral drug resistance between subtype B HIV-1 and several group M non-B subtypes. Non-B subtypes, e.g. subtype C HIV-1 variants, are known to possess naturally occurring polymorphisms at several RT and PR codons that are implicated in drug resistance [20, 21]. In some studies, the presence of these polymorphisms did not significantly reduce susceptibility to ARVs in phenotypic resistance assays or limit the effectiveness of an initial antiretroviral therapy regimen for a period of up to 18 months [20, 22]. However, it has also been suggested that polymorphisms at resistance positions may sometimes facilitate selection of novel pathways leading to drug resistance, especially with incompletely suppressive antiretroviral regimens [20]. This, in turn, may have important clinical implications with respect to choice of effective antiretroviral therapy (ART). This warrants increased genotypic surveillance on a worldwide basis, as the prevalence of non-B HIV-1 infection is increasing rapidly.
As illustrated in Figures 12.1–12.5, it has been possible to select numerous drug resistance mutations for all licensed ARVs and investigational agents such as some HIV-1 entry inhibitors [5, 6, 23]. In view of the hypervariability of HIV-1 and the inability of many current antiretroviral combinations to completely suppress viral replication, especially in patients with prior treatment failure, it is essential that new anti-HIV drug discovery initiatives focus on the identification of new therapeutic targets and the development of ARVs with more robust genetic barriers for resistance and a broader spectrum of activity against drug-resistant HIV-1 variants.
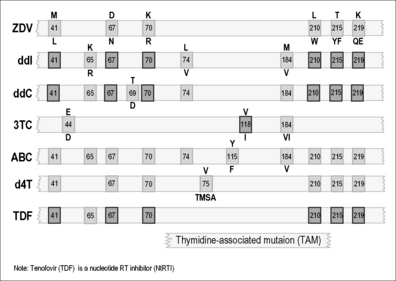
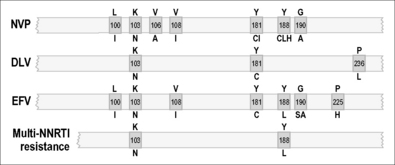

Figure 12.3 Common PR mutations associated with HIV drug resistance.
The letter designation at the top of a box refers to the amino acid that is present in the wild-type sequence of PR. The letters at the bottom refer to substitutions associated with drug resistance. Sometimes, several different amino acid changes at the same codon can confer drug resistance, as for example both I and L in the case of position [46]. Resistance to darunanavir and tipranavir involve multiple combinations of the above mutations, and both these drugs have a higher genetic barrier for resistance then other members of the PI family of drugs.
Generation of HIV-1 Drug Resistance
Resistance mutations to ARVs may arise spontaneously as a result of the error-prone replication of HIV-1 and, in addition, are selected both in vitro and in vivo by pharmacological pressure [24–26]. The high rate of spontaneous mutation in HIV-1 has been largely attributed to the absence of a 3’-5’exonuclease proof-reading mechanism. Sequence analyses of HIV-1 DNA have detected several types of mutations including base substitutions, additions, and deletions [24]. The frequency of spontaneous mutation for HIV-1 varies considerably as a result of differences among viral strains studied in vitro [27]. Overall mutation rates for wild-type laboratory strains of HIV-1 have been reported to range from 97 × 10-4 to 200 × 10-4 per nucleotide for the HXB2 clonal variant of HIV-1 to as high as 800 × 10-4 per nucleotide for the HIV-1 NY5 strain [24, 27].
In addition to the low fidelity of DNA synthesis by HIV-1 RT, other interdependent factors that affect rates of HIV mutagenesis include RT processivity, viral replication capacity, viral pool size, and availability of target cells for infection [28–31]. It follows that an alteration in any or a combination of these factors might influence the development of HIV drug resistance. There are also data showing that thymidine analog mutations (TAMs) in RT can significantly increase the likelihood of further mutational HIV-1 distributions and evolution of drug resistance; furthermore, this can happen in the presence or absence of concomitantly administered NRTIs [32, 33].
Inhibitors of Reverse Transcriptase
The RT enzyme is encoded by the HIV pol gene and is responsible for the transcription of double-stranded proviral DNA from viral genomic RNA. Two categories of drugs have been developed to block RT; these are NRTIs that act to arrest DNA chain elongation by acting as competitive inhibitors of RT and NNRTIs that act as non-competitive antagonists of enzyme activity by binding to the catalytic site of RT. NRTIs are administered to patients as precursor compounds that are phosphorylated to their active triphosphate form by cellular enzymes. These compounds lack a 3’ hydroxyl group (OH) necessary for elongation of viral DNA. These analogs can compete effectively with normal deoxynucleotide triphosphate (dNTP) substrates for binding to RT and incorporation into viral DNA [34, 35].
NNRTI antiviral activity involves the binding of these non-competitive inhibitors to a hydrophobic pocket close to the catalytic site of RT [36, 37]. NNRTI inhibition reduces the catalytic rate of polymerization without affecting nucleotide binding or nucleotide-induced conformational change [38]. NNRTIs are particularly active at template positions at which the RT enzyme naturally pauses. NNRTIs do not seem to influence the competition between dideoxynucleotide triphosphates (ddNTPs) and the naturally occurring dNTPs for insertion into the growing proviral DNA chain [39].
Both types of RT inhibitors have been shown to successfully diminish plasma viral burden in HIV-1-infected subjects. However, monotherapy with all drugs has led to drug resistance. Patients who receive combinations of three or more drugs are less likely to develop resistance, since these “cocktails” can suppress viral replication with much greater efficiency than single drugs or two drugs in combination. Although mutagenesis is less likely to happen in this circumstance, it can still occur, and the emergence of drug-resistant breakthrough viruses has been demonstrated in patients receiving highly active ART [40, 41]. Furthermore, the persistence of reservoirs of latently infected cells represents another major impediment to currently used anti-HIV chemotherapy [42]. Replication of HIV might resume once therapy is stopped or interrupted and, therefore, eradication of a latent reservoir of 105 cells might take as long as 60 years, a goal that is not practical with currently available drugs and technology [42, 43].
Mutations associated with drug resistance have been reported in response to the use of any single NRTI or NNRTI [44]. However, not all drugs elicit the same mutagenic response; sensitivity and resistance patterns must be considered on an individual drug basis. For example, patients on 3TC monotherapy may develop high-level, i.e. 1000-fold resistance within weeks, whereas 6 months or more are often required in order for sensitivity to ZDV to drop by 50- to 100-fold. In contrast, HIV may appear to remain partially sensitive, even after prolonged monotherapy, to several other commonly used nucleoside analogs: ddI (didanosine), d4T (stavudine), and ABC (abacavir). Of the latter, only ABC is commonly used in Western countries today, although the use of d4T, which is often toxic, continues in many developing country settings, mainly because it is inexpensive. In the case of ZDV, increases in IC50 below threefold are regarded as non-significant, while 10-to 50-fold increases usually represent partial resistance, and increases above 50-fold denote high-level resistance.
Patient resistance to nucleoside analogs can often develop independently of the dose of drug that is administered. Tissue culture data have shown that HIV-1 resistance can be easily demonstrated against each of NRTIs, NNRTIs, and PIs, by gradually increasing the concentration of compound in the tissue culture medium [45, 46]. Cell lines are especially useful in this regard, since HIV replication occurs very efficiently in such hosts. Tissue culture selection provides an effective pre-clinical means of studying HIV mutagenesis, especially since the same resistance-conferring mutations that arise in cell culture are usually predictive of those that will appear clinically. Owing to the high turnover and mutation rate of HIV-1, the retroviral quasispecies will also include defective virions and singly mutated drug-resistant variants that are present prior to commencement of therapy. Multiply mutated variants appear later, because it requires time to accumulate multiple mutations within a single viral genome. Therefore, multiply mutated viruses are not commonly found in the retroviral pool of untreated patients. An exception to this involves cases of new infection with drug-resistant viruses transmitted from extensively treated individuals. Patients with advanced infection have a higher viral load and a broader range of quasispecies than newly infected individuals. Such patients are often immunosuppressed and may also have diminished ability to immunologically control viral replication, possibly leading to more rapid development of drug resistance.
Site-directed mutagenesis has shown that a variety of RT mutations encode HIV resistance to both NRTIs and NNRTIs. Crystallographic and biochemical data have demonstrated that mutations conferring resistance to NNRTIs are found in the peptide residues that make contact with these compounds within their binding pocket [36, 37].
Resistance-encoding mutations to NRTIs are found in different regions of the RT enzyme, probably due to the complexities of nucleoside incorporation, which involve several distinct steps. These mutations can decrease RT susceptibility to nucleoside analogs. A summary of primary RT mutations has been published [44].
It has also been shown that a family of insertion and deletion mutations between codons 67 and 70 can cause resistance to a variety of NRTIs including ZDV, 3TC, ddI, ddC, and d4T. Usually, these insertion mutations confer multidrug resistance (MDR) when present in a ZDV-resistant background. Another less frequently observed resistance mutation, K65R, has been shown to be associated with prior treatment with ABC- and tenofovir (TDF)-containing regimens and results in reduced antiviral susceptibility to both of these agents. Hence, resistance to these ARVs can develop via genetic pathways involving either the TAMs or K65R as hallmark drug resistance mutations [47]. In recent years, the proportion of genotyped clinical samples containing K65R has increased from less than 1 to almost 4%, reflecting the increased use of TDF in first-line treatment regimens in developed countries.
Diminished sensitivity to NNRTIs appears quickly both in culture selection protocols and in patients [36, 48]. NNRTIs share a common binding site, and mutations that encode NNRTI resistance are located within the binding pocket that makes drug contact [36, 37, 39–46, 48–53]. This explains the finding that extensive cross-resistance is observed among several approved NNRTIs. A substitution at codon 181 (tyrosine to cysteine) (Y181C) is a common mutation that encodes cross-resistance among many NNRTIs [49, 54, 55]. Replacement of Y181 by a serine or histidine also conferred HIV resistance to NNRTIs [56]. A mutation at amino acid 236 (proline to leucine) (P236L), conferring resistance to a particular class of NNRTIs that include delavirdine, can also diminish resistance to nevirapine (NVP) and efavirenz (EFV), particularly if a Y181C mutation is also present in the same virus [57]. Other important substitutions are Y188C and Y188H and can also confer NNRTI resistance.
Another drug resistance mutation, namely K103N (lysine to asparagine), is commonly observed and is responsible for reduced susceptibility to many approved NNRTIs including efavirenz and nevirapine [49, 54, 55]. Substitution of K103N results in alteration of interactions between NNRTIs and RT. The K103N mutation shows synergy with Y181C in regard to resistance to NNRTIs, unlike antagonistic interactions involving Y181C and P236L [55]. Recently, a mutation at position E138K was shown to encode resistance against a newer NNRTI termed etravirine that has been shown to be effective in second-line therapeutic regimens [58]. This E138K mutation also seems to be associated with resistance to rilpivirine, a novel NNRTI that is currently undergoing advanced clinical trials for potential use in first-line therapy in association with NRTIs.
Resistance to NNRTIs is also observed in cell-free enzyme assays [52, 54, 59–61]. Both Y181I and Y188L mediate decreased sensitivity to NNRTIs without affecting either substrate recognition or catalytic efficiency, supporting the idea that resistance to NNRTIs is attributable to diminished ability of these drugs to be bound by RT.
In recent years, a new member of the NNRTI family, etravirine (ETV), has been approved for use in patients with pre-existing resistance against NVP and EFV. ETV possesses excellent activity against viruses containing the K103N and T181C mutations, among others, that are associated with resistance against first-generation NNRTIs [62, 63]. However, the activity of ETV may be compromised by other mutations such as E138K, although it remains to be determined whether this mutation will appear to significant extent in patients who fail ETV-containing ARV regimens [64]. Several drug companies are now trying to develop novel NNRTIs that will hopefully have a higher genetic barrier for resistance than either NVP or EFV for use in first-line ARV regimens. Several such compounds are now being studied in clinical trials.
Protease Inhibitors
Drug-resistant viruses have been observed in the case of all PIs developed to date [65–67]. In addition, some strains of HIV have displayed cross-resistance to a variety of PIs after either clinical use or in vitro drug exposure [65–67]. In general, the patterns of mutations observed with PIs are more complex than those observed with RT antagonists. First, a greater number of mutations within the PR gene are required for resistance to occur. This is especially true for both darunavir and tipranavir, that almost always require a greater accumulation of the resistant mutations listed in Figure 12.3 in order for resistance to occur, than would be the case for any other member of the PI family of drugs. There is also greater variability, among PIs, in temporal patterns of appearance of different mutations and the manner in which different combinations of mutations can give rise to phenotypic resistance. These data suggest that the PR enzyme can adapt more easily than RT to pressures exerted by antiviral drugs. At least 40 mutations in PR have been identified as responsible for resistance to PIs [65–67].
Certain of the mutations within the PR gene are more important than others and can confer resistance, virtually on their own, to at least certain PIs [65–67]. One mutation, in particular, D30N, is probably unique to nelfinavir (NFV), a potent HIV PI. However, a variety of other mutations may confer cross-resistance among multiple drugs within the PI family. In addition, wide arrays of secondary mutations that, when combined with primary mutations, can cause increased levels of resistance to occur have been observed. On the other hand, the presence of certain of these secondary mutations on their own may not lead to drug resistance, and, in this context, some of these amino acid changes should be considered to represent naturally occurring polymorphisms. In addition, it should be noted that resistance to PIs can also result from mutations within the substrates of the PR enzyme, i.e. the gag and gag-pol precursor proteins of HIV. A variety of studies have now shown that mutations at cleavage sites within these substrates can be responsible for drug resistance, both in tissue culture and in treated patients. However, the full clinical significance of cleavage site mutations in regard to PR resistance is not understood.
Fusion and Entry Inhibitors
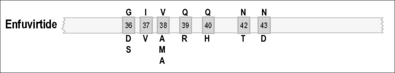
ENF is a 36 amino acid synthetic peptide that structurally mimics the heptad repeat 2 (HR2) region of gp41. It also inhibits the association of HR2 with the heptad repeat (HR1) of the same protein. This action impedes the formation of a six-helix bundle that mediates the fusion of the HIV envelope and cell membrane. ENF is generally reserved for highly experienced patients who have failed all other therapies and is used in association with an optimized background ARV regimen [68].
Mutations at positions 36–45 (GIVQQQNNLL) of the HR1 region are extremely important in the development of resistance. Even a single amino acid substitution can cause a significant reduction in drug susceptibility. As a result, ENF is considered to have a low genetic barrier for resistance. The emergence of mutations at amino acids 36–45 was detected in 94% (205/218) of viruses from patients experiencing virological failure while taking ENF in each of two clinical trials. The most frequent mutations were at positions 38 (47.7%, 104/218; most often V38A), 43 (30.7%, 58/218; most often N43D), and 36 (26.6%, 58/218; most often G36D) [68–70].
MVC is a CCR5 co-receptor inhibitor. It binds to the CCR5 co-receptor of the susceptible host cell. By doing so, it prevents the HIV-1 gp120 protein from interacting with the CCR5 co-receptor, resulting in blockage of viral entry. The results of the MOTIVATE trials showed that MVC can be effective in the treatment of advanced HIV disease, when combined with other ARVs, in patients whose viruses were CCR5-tropic. MVC seems not to be effective against viruses that are tropic for the CXCR4 co-receptor. Its use is limited to patients who have a laboratory assay that confirms that their virus is CCR5-tropic. Most testing for MVC eligibility is now conducted by sequencing of the HIV-1 env gene, although a phenotypic test is also available for this purpose [71, 72].
Resistance to MVC is associated with both a complex array of mutations in the V3 loop that do not cause cross-resistance to ENF as well a pre-existing minority populations of CXCR4-tropic viruses that may became predominant after treatment with MVC [73, 74].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


