Chapter 34 Congenital Abnormalities
Definition and causes





Chromosome and gene abnormalities
Genetic disorders (Mendelian inheritance)



Teratogenic causes
A teratogen is any agent that raises the incidence of congenital abnormality. The list of known and suspected teratogens is continually growing (Box 34.1).
Box 34.1 Known and suspected teratogens
Multifactorial causes
These stem from a genetic defect in addition to one or more teratogenic influences.







Gastrointestinal malformations
Most of the abnormalities affecting this system call for prompt surgical intervention.
Gastroschisis and exomphalos
Gastroschisis (Fig. 34.1) is a paramedian defect of the abdominal wall with extrusion of bowel that is not covered by peritoneum.
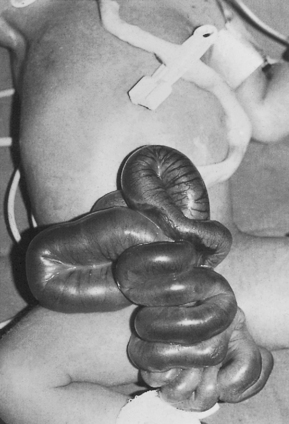
Fig. 34.1 Gastroschisis showing prolapsed intestine to the right of the umbilical cord.
(From Rennie & Roberton 1999, with permission of Churchill Livingstone.)
Immediate management of both is as follows:




Atresias
Oesophageal atresia







Cleft lip and cleft palate
Cleft lip may be unilateral or bilateral and is very often accompanied by cleft palate.



Abnormalities relating to respiration
Diaphragmatic hernia




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


