Cardiovascular disease remains the leading cause of mortality in the United States. In 2008, the overall rate of death attributable to cardiovascular disease (CVD) was 244.8 per 100 000.1 From 1998 to 2008, the rate of death attributable to CVD declined by 30%.1 In the United States, a person has a cardiac event approximately every 25 seconds, and every minute someone dies from a cardiac event.1 CVD causes 1 in 3 deaths in the United States, and 1 in 9 death certificates mentioned heart failure; 1 in 3 people had some form of CVD.1 In 2009, 4.2 million cardiovascular operations and procedures were performed on men and 3.3 million on women. Between 2010 and 2030, it is expected that the total medical costs of CVD will triple from 273 billion to 818 billion dollars.1 The critical care nurse is in a unique position to assist with educating the public on a daily basis about CVD and the risk factors that are modifiable with changes in habits or lifestyles. An understanding of the pathology of CVD processes and clinical management allows the critical care nurse to accurately anticipate and plan interventions. This chapter focuses on cardiac disorders commonly seen in the critical care environment. The biggest contributor to cardiovascular system–related morbidity and mortality is coronary artery disease (CAD). Atherosclerosis is a progressive disease that affects arteries throughout the body. In the heart, atherosclerotic changes are clinically known as CAD. This disease process is also known by the term coronary heart disease (CHD) because other heart structures ultimately become involved in the disease process. The atherosclerotic vascular changes that lead to CAD may begin in childhood. Research and epidemiologic data collected during the past 50 years have demonstrated a strong association between preventable and nonpreventable risk factors and the development of CAD.1,2 These risk factors are further delineated as nonmodifiable and modifiable coronary risk factors (Box 15-1). The severe effects of CAD occur as a person ages. In general, CAD symptoms are seen in persons age 45 years and older.1 Traditionally, CAD has been regarded a primarily male disease. Research is, however, showing that it affects women as well.1–3 Men typically develop external manifestations of the disease about 5 to 10 years earlier compared with women. The prevalence of cardiovascular disease is higher among women starting at age 75 years.1–3 CAD rates for postmenopausal women are two to three times higher than those for premenopausal women of the same age.1 Primary cardiovascular risk factors are different in men and women, with women having higher rates of diabetes and hypertension compared with men.1 A positive family history is one in which a close blood relative has had a myocardial infarction (MI) or stroke before age 60 years. This family history suggests a genetic or lifestyle predisposition to the development of CAD. Individuals with a family history had a 50% greater risk of having an acute MI as demonstrated in the INTERHEART study, a large study that looked at risk factors of patients in 52 countries who experienced an MI.4 Hyperlipidemia is a leading factor responsible for severe atherosclerosis and the development of CAD. Millions of adults above age 20 years have total serum cholesterol levels above 240 milligrams per deciliter (mg/dL). Assessing total serum cholesterol and triglyceride levels is essential to the assessment of cardiovascular risk in patients.1 A lipid panel blood test will measure the following values: • High-density lipoprotein (HDL) cholesterol • Low-density lipoprotein (LDL) cholesterol • Very-low–density lipoprotein (VLDL) cholesterol Treatment of hyperlipidemia has advanced beyond the concept of lowering total cholesterol to treatment of specific lipoprotein abnormalities.1,3,5 The target levels for specific serum lipids are listed in Table 15-1. TABLE 15-1 LIPID GUIDELINES AND RISK FOR CORONARY ARTERY DISEASE HDL, High-density lipoprotein; LDL, low-density lipoprotein; VLDL, very-low–density lipoprotein. *Values outside the target range increase the risk for coronary artery disease. HDL cholesterol is frequently described as the “good cholesterol” because higher serum levels exert a protective effect against acute atherosclerotic events. All the reasons are not completely understood, but one recognized physiologic effect is the ability of HDL to promote the efflux of cholesterol from cells. This process may minimize the accumulation of foam cells in the arterial wall and decrease the risk of developing atherosclerosis.6 High HDL cholesterol levels confer anti-inflammatory and antioxidant benefits on the arterial wall.6 In contrast, a low HDL cholesterol level is an independent risk factor for the development of CAD and other atherosclerotic conditions. HDL cholesterol is generally higher in women, and levels can be raised by physical exercise and by stopping smoking. In patients with low HDL cholesterol levels, when lifestyle changes are ineffective, the HDL level can be raised by medications such as extended-release nicotinic acid (niacin) and fibrates. LDL cholesterol is usually described as the “bad cholesterol” because high levels are associated with an increased risk of acute coronary syndrome (ACS), stroke, and peripheral arterial disease (PAD). High LDL levels initiate the atherosclerotic process by infiltrating the vessel wall and binding to the matrix of cells beneath the endothelium.6 LDL cholesterol also exerts an inflammatory effect on the arterial vessel wall.6 A high LDL cholesterol level is initially managed by nonpharmacologic lifestyle changes, such as weight loss, smoking cessation, low-fat diet, physical exercise, and attainment of a normal body size as measured by the body mass index (BMI). If lifestyle changes are insufficient to reduce the LDL cholesterol level in the bloodstream, the medication category of choice is a statin. Numerous research studies have conclusively demonstrated that lowering the LDL cholesterol with statins for primary or secondary prevention is highly effective in lowering mortality from CAD.1,3 Therapy should be tailored to treat the individual cardiovascular risk profile. The target LDL cholesterol is determined according to the individual’s risk profile as described in Table 15-1. Triglycerides are serum lipids that constitute an additional atherogenic risk factor. A fasting triglyceride level above 150 mg/dL in adults is considered elevated and is a risk factor for heart disease and stroke. Recent guidelines suggest that triglyceride levels below 100 mg/dL may be optimal.7 The mean level of triglycerides for American adults ≥20 years of age is 137.6 mg/dL.1 LDL cholesterol can be further analyzed by the category of lipid particles that make up the total LDL value. Researchers have investigated the function of several lipid particles to determine their role in the development of premature atherosclerotic CAD. One particle that has been extensively studied is lipoprotein(a), which is abbreviated Lp(a) and described verbally as “LP little a.” Lp(a) is manufactured in the liver and circulates in the bloodstream bound to a large glycoprotein called apolipoprotein(a), abbreviated as apo(a).8 The Lp(a)–apo(a) lipid particle concentration is elevated in the presence of inflammation, and it stimulates atheroma and clot formation in inflamed arteries.8 This effect is thought to occur because the apo(a) is structurally similar to plasminogen, a protein essential for clot formation. Lp(a) levels are 90% genetically determined. Elevated Lp(a) plasma levels constitute the most frequently encountered genetic lipid disorder in families with premature CAD.8 Testing for Lp(a) is reserved for high-risk patient populations, such as those with a strong family history of premature atherosclerotic disease and for patients with premature CAD who do not exhibit the expected cardiac risk factors. Reduction of Lp(a) levels to below 30 mg/dL is the therapeutic goal. This usually is achieved by ingesting high doses (1500 to 2000 mg/day) of extended-release nicotinic acid (niacin). The Lp(a) level is not reduced by statins, medications that traditionally lower LDL levels, or by physical exercise, a low-fat diet, weight loss, or tight control of blood glucose levels.8 Lifestyle changes are recommended, and research is ongoing, but a cure has not yet been found. A diet rich in saturated fats leads to elevated cholesterol levels in the blood. The first line of treatment to lower elevated serum cholesterol is a low-fat, high-fiber diet and increased physical exercise.9,10 If these measures are not effective, lipid-lowering medications are indicated. This approach sounds simple, but less than one half of the people who qualify for lipid reduction therapy are taking their medications; only one third of treated patients reach their LDL target. Obesity is a disease of modern times. A body mass index (BMI) of greater than 30 kilograms per meter squared (kg/m2) is considered obese. Global estimates are more than 1 billion overweight adults, and at least 300 million of these people are obese. Obesity is often associated with a sedentary lifestyle accompanied by the calories consumed and portion sizes. A high risk of coronary heart disease is among the well-established adverse health effects associated with excess weight. Hypertension, hypercholesterolemia, and diabetes are among the clinical conditions that are important mediators of this association.1–3 Currently, one third of the adult population in the United States is obese.1 The BMI is a mathematic formula used to assess body weight relative to height. BMI is used to evaluate the threat of excess weight as a risk factor for CAD and permits comparisons of people of different gender, age, height, and body type.1 BMI is calculated as the weight in kilograms divided by the square of the height in meters (kg/m2). The BMI calculation in metric units is shown in Box 15-2. A normal BMI is between 18.5 and 25 kg/m2. A BMI between 25 and 30 kg/m2 indicates that the person is overweight. A BMI greater than 30 kg/m2 is the definition of obesity.1 Regular vigorous physical activity using large muscle groups promotes physiologic adaptation to aerobic exercise, which can prevent the development of CAD and reduce symptoms in patients with established cardiovascular disease.11 Exercise also reduces the incidence of many other diseases, including type 2 diabetes, osteoporosis, obesity, depression, and cancers of the colon and breast.11 Many research trials have demonstrated the positive effects of physical activity on the other major cardiac risk factors.11 Exercise alters the lipid profile by decreasing LDL cholesterol and triglyceride levels and increasing HDL cholesterol levels.11 Exercise reduces insulin resistance at the cellular level, lowering the risk for developing type 2 diabetes, especially if combined with a weight-loss program.11 Epidemiologic studies indicate that participating in physical athletics in one’s youth does not confer protection in later years. A sedentary lifestyle has negative effects, regardless of age, gender, BMI, smoking status, presence or absence of hypertension, or abnormal lipoprotein profile. Lifelong physical activity is necessary to prevent atherosclerotic CAD and stroke. Hypertension is often described as the “silent killer” because 30% of those affected are unaware they have seriously elevated blood pressure.1 In the United States, 1 person in 3 has hypertension. Normal blood pressure is described as a systolic blood pressure (SBP) below 120 mm Hg and a diastolic blood pressure (DBP) below 80 mm Hg. Hypertension is defined as an SBP greater than 140 mm Hg or DBP greater than 90 mm Hg. Controlled hypertension is a term that describes maintaining blood pressure within the normal range with the use of antihypertensive medications.1 It is essential that patients understand that sustained elevation of blood pressure leads inexorably to atherosclerosis, heart failure, kidney failure, stroke, and heart attack.12 Hypertension is so widespread in industrialized societies that even a normotensive person at age 55 years has a 90% lifetime risk of developing hypertension. This implies that even normotensive persons should adopt interventions to maintain a normal blood pressure.1 Hypertension is a cardiac risk factor because high SBP damages the arterial endothelium, leading to vascular inflammation that encourages the formation of plaque. Hypertension is a complex, multifactorial disease process, which is divided into stages for the purposes of treatment, as shown in Table 15-2. TABLE 15-2 BLOOD PRESSURE GUIDELINES AND RISK FOR CORONARY ARTERY DISEASE BP, Blood pressure; CAD, coronary artery disease. *Values greater than normal increase the risk for CAD and heart failure. Prehypertension is an SBP of 120 to 139 mm Hg or DBP above 85 mm Hg.1,11,12 Hypertension is diagnosed when the blood pressure is above 140/90 mm Hg. Hypertension affects another one in three adults in the United States. After the blood pressure is above 140/80 mm Hg, hypertension is described as stage 1 or stage 2 (see Table 15-2). Projections show that by 2030, an additional 27 million people could have hypertension.1 The current guidelines recommend the goal of treatment for the person with hypertension without other risk factors is to achieve a blood pressure below 140/80 mm Hg. For the person with hypertension who already has diabetes or kidney disease, the target blood pressure is below 130/80 mm Hg. A normal blood pressure is below 120/80 mm Hg.11,12 Lifestyle interventions that can normalize blood pressure include physical exercise, a low-salt diet, limiting alcohol intake, and achieving normal body weight. Most patients are started on a diuretic, and if this is insufficient, they may be placed on an angiotensin-converting enzyme inhibitor (ACEI), angiotensin receptor blocker (ARB), beta-blocker, or calcium channel blocker. Most patients require at least two medications, one each from different medication classifications, to normalize their blood pressure.12 Hypertensive emergencies with acute organ damage are discussed in the last section of this chapter. The greater the number of cigarettes smoked per day, the greater is the risk of developing CAD, acute MI, and stroke.1 Cigarette smoking unfavorably alters serum lipid levels, decreases HDL cholesterol level, and increases LDL cholesterol and triglyceride levels. Smoking increases the risk of CHD at all levels, including less than five cigarettes per day.1 Smokers are two to four times more likely to develop CAD compared with nonsmokers.1 Passive, secondhand smoke exposure also increases cardiovascular risk for nonsmoking adults.1,13 Nonsmokers who are exposed to secondhand smoke at home or at work increase their risk of developing coronary heart disease by 30%.1 Current cigarette smoking is a powerful predictor for the development of unstable angina, and myocardial infarction.1,14,15 Within 1 year of giving up cigarettes, an ex-smoker’s risk of developing CAD decreases significantly. Nicotine is addictive, and giving up smoking is difficult. People need tremendous support to be able to “kick the habit.” The good news is that many do quit, and smoking in the United States continues to decline.1 Chapter 20 provides patient education guidelines on how to stop smoking. Individuals with diabetes mellitus (types 1 and 2) have a higher incidence of CHD compared with the general population. Elevated blood glucose level is a known risk factor for development of vascular inflammation associated with atherosclerosis. For decades, the diagnosis of diabetes was based on plasma glucose criteria. In 2009, the American Diabetes Association recommended the use of the hemoglobin (Hgb) A1C test to diagnose diabetes, with at threshold of 6.5% or greater, which was adopted as standard practice in 2010.16 The criteria for the diagnosis of diabetes is: A1C 6.5% or greater, a fasting blood glucose 126 mg/dL or greater, or 2-hour plasma glucose 200 mg/dL or greater during an oral glucose tolerance test.16 Or, in a patient with classic symptoms of hyperglycemia a random plasma glucose above 200 mg/dL signifies diabetes. 16 A fasting blood glucose concentration between 100 to 125 mg/dL or an A1C of 5.7% to 6.4% represents an increased risk for diabetes (Table 15-3). The upper limit for a normal fasting plasma glucose level is 125 mg/dL.16,17 Patients with diabetes have an increased risk of developing CAD and have worse clinical outcomes after ACS events.16,17 In a multinational study of patients who were seen at hospitals with symptoms of ACS, almost 1 in 4 had a known history of diabetes.18 More detailed information on type 2 diabetes and the use of insulin and oral medications to control blood glucose levels and combat insulin resistance is included in Chapters 32 and 33. TABLE 15-3 FASTING BLOOD GLUCOSE AND RISK FOR CORONARY ARTERY DISEASE *Values greater than normal increase the risk for CAD and kidney failure. Chronic kidney disease is considered a risk equivalent for CAD.1–3,19 This means that patients with chronic kidney disease have as much risk of experiencing a coronary event as if they already had CAD.1–3,20 The risk of death for the patient with acute MI rises as the serum creatinine level increases.20 Metabolic syndrome refers to the clustering of risk factors associated with CVD and type 2 diabetes.1 About one third of people in the United States have metabolic syndrome. The following are risk factors1,21: • Fasting plasma glucose above 100 mg/dL or taking medications to lower elevated blood glucose. • HDL cholesterol below 40 mg/dL in men and below 50 mg/dL in women, or taking medications to raise HDL levels. • Triglycerides above 150 mg/dL or taking medications to lower elevated triglycerides. • Waist circumference above 40 inches (102 cm) in men, or above 35 inches (88 cm) in women. • Blood pressure (BP) above 130 mm Hg systolic or above 85 mm Hg diastolic, or taking antihypertensive medications. Substantial progress has been made in the awareness, treatment, and prevention of CVD in women.22 In 2007, CVD still caused approximately one death per minute among women in the United States.22 After age 65 years, a higher percentage of women than men have hypertension. Average body weight continues to increase, with nearly 2 out of every 3 women in the United States above age 20 years now being overweight or obese.22 The average age for the first acute MI in men is 65.8 years, and in women, it is 70.4 years.1 The incidence of CVD is two to three times higher among postmenopausal women than among women who are premenopausal.1 In the past, it seemed logical to prescribe hormone replacement therapy (HRT) to treat the symptoms of menopause.22 Current 2011 guidelines do not recommend the use of hormone therapy for primary or secondary prevention of CVD.22 Data from the Framingham Heart Study indicate the lifetime risk for cardiovascular disease is more than one in two for women at age 40 years, and in the 2007 update of Guidelines for the Prevention of CVD in Women, a new algorithm for risk classification in women was adopted that stratified women’s risk into 3 categories: • At high risk: documented CAD or CVD risk equivalents, such as the presence of documented CVD, diabetes mellitus, end stage or chronic kidney disease, or 10-year predicted risk for CHD>20% • At risk: given the presence of subclinical vascular disease or poor exercise tolerance on treadmill testing • At optimal risk: in the setting of a Framingham risk score less than 10%, absence of major CVD risk factors, and engagement in a healthy lifestyle.1,22 In 2011, these guidelines added an acknowledgement of several 10-year risk equations for the prediction of 10-year global CVD risk, such as the updated Framingham CVD risk profile and the Reynolds risk score for women.22 • Absence of clinical CVD and the presence of all ideal levels of total cholesterol (<200 mg/dL) • Blood pressure (<120/80 mm Hg) • Fasting blood glucose (<100 mg/dL) • Adherence to healthy behaviors • Lean body mass index (<25 kg/m2) • Participation in physical activity at recommended levels • Pursuit of eating pattern as suggested by Dietary Approaches to Stop Hypertension (DASH diet)23 Cardiovascular disease kills more than 500,000 women annually in the United States. Mortality rates for women after an acute MI are higher than for men: 38% compared with 25%. Risk factors associated with acute MI more strongly in women than in men include hypertension, diabetes mellitus, alcohol intake, and physical inactivity.22,23 Many reasons contribute to higher mortality from acute MI in women, and these include waiting longer to seek medical care, having smaller coronary arteries, being older when symptoms occur, and experiencing very different symptoms from those of men of the same age.24,25,26 The inflammatory marker most frequently cited is C-reactive protein (CRP). It is measured as high-sensitivity CRP (hs-CRP).27–29 CRP is associated with an increased risk of development of other cardiovascular risk factors including diabetes, hypertension, and weight gain.28–30 The higher the hs-CRP value, the greater is the risk of a coronary event, especially if all other potential causes of systemic inflammation such as infection can be ruled out. Value ranges for hs-CRP are shown in Table 15-4. If other systemic inflammatory conditions such as bronchitis or urinary tract infection are present, the hs-CRP test loses all predictive value.28,29 CRP and other inflammatory markers are used to estimate the probability of future acute coronary events.29,30 During ACS events, there is widespread activation of neutrophils in the cardiac circulation (measured from the coronary sinus), which suggests that inflammation is not limited to one unstable plaque.31 TABLE 15-4 C-REACTIVE PROTEIN AND RISK FOR CORONARY ARTERY DISEASE *Values above 1 mg/dL increase the risk for CAD, but test results are not valid in the presence of infection or other inflammatory condition. Normal values may vary slightly between clinical laboratories; however, values below 1 mg/dL are usually considered normal. A test result greater than 10 mg/L suggests a noncoronary source of inflammation or infection. Based on data from Pearson TA, et al. Markers of inflammation and cardiovascular disease, application to clinical and public health practice: a statement for healthcare professionals from the Centers for Disease Control and Prevention and the American Heart Association. Circulation. 2003;107(3):499. Certain medical conditions are risk equivalents of CAD. A risk equivalent means the person has the same risk of having an acute MI as if he or she had coronary heart disease already. Two noncardiac medical conditions considered risk equivalents for CAD: diabetes mellitus and chronic kidney disease.30 PAD and cerebral vascular disease are atherosclerotic conditions that are also considered CAD risk equivalents.30 CAD has multifactorial causation; the greater the number of risk factors, the greater the risk of developing CAD.1,3,30,32 The best time for an individual to make lifestyle changes is before the symptoms of CAD occur. Patients with two or more risk factors or with one or more of the CAD risk-equivalent diseases have the greatest potential to benefit from risk factor reduction and lifestyle change.30 The major risk factors for developing CAD have been extensively documented in large epidemiologic studies: smoking, family history, adverse lipid profile, and elevated blood pressure. If a person has symptoms of CAD or has previously had an ACS event, the goal of any lifestyle change or medication is called secondary prevention, or preventing another heart attack.11 If an individual matches the risk profile described previously but does not have symptoms of CAD or has not had an acute MI, the treatment plan is described as primary prevention. The constellation of cardiac risk factors is well established and can predict development of CAD for most populations in the developed world.30 Atherosclerosis is a chronic inflammatory disorder that is characterized by an accumulation of macrophages and T lymphocytes in the arterial intimal wall. A high LDL cholesterol concentration is one of the triggers of vascular inflammation. The inflammation injures the wall, allowing the LDL cholesterol to move into the vessel wall below the endothelial surface.6 Blood monocytes adhere to endothelial cells and migrate into the vessel wall. Within the artery wall, some monocytes differentiate into macrophages that unite with and then internalize LDL cholesterol. The foam cells that result are the marker cells of atherosclerosis.6 Elevated LDL cholesterol levels promote low-level endothelial inflammation, which allows lipoproteins to infiltrate the intimal vessel wall. After it has infiltrated under the endothelium, LDL cholesterol tends to stay within the vessel wall rather than return to the circulation. This contrasts with the actions of HDL cholesterol, which enters the vessel wall, helps efflux cholesterol from cells, and then returns to the circulation.6 The actions of HDL cholesterol may help minimize the number of foam cells in the artery wall.6 When a mature atherosclerotic plaque develops, it is not uniform in composition. It has a lipid liquid center filled with procoagulant factors. A connective tissue fibrous cap covers the top of the fluid lipid center.29,31 The abrupt rupture of this cap allows procoagulant lipids to flood into the vessel lumen and rapidly form a coronary thrombosis, as shown in Table 15-5. As the enlarging clot blocks blood flow through the coronary artery, a “heart attack” will occur unless adequate collateral circulation from other coronary vessels occurs. Symptoms and suggested cardiac interventions at appropriate stages in development of CAD are listed in Table 15-5. TABLE 15-5 TIMELINE OF ATHEROGENESIS DEVELOPMENT DEPICTED BY LONGITUDINAL SECTION OF AN ARTERY Illustration modified from Antman EM, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction—executive summary. Circulation. 2004;110:588. Plaques that are likely to rupture are saturated with macrophages and other inflammatory cells. These vulnerable plaques are usually not obstructive and are situated at bends or branch points in the arterial tree.3 It is not known what factors cause the fibrous cap to rupture or erode. As deep fissures in the cap expose the procoagulant factors to the blood plasma, an unstoppable cycle is put into motion. When platelets in the bloodstream are exposed to collagen, necrotic debris, von Willebrand factor, and thromboxane, a clot is formed and can occlude the coronary artery. Highly fibrotic plaques do not rupture. The type of atherosclerotic plaque that is prone to rupture has a weak fibrous cap and a large amount of liquid cholesterol within the core (see Table 15-5). A reduction in blood cholesterol decreases atherosclerotic plaque size by decreasing the amount of liquid cholesterol within the plaque core.9 Lowering cholesterol levels does not change the dimensions of the fibrous or calcified portions of the plaque. However, lower cholesterol levels reduce vascular inflammation and make vulnerable plaque less likely to rupture. The term acute coronary syndrome (ACS) is used to describe the array of clinical presentations of CAD that range from unstable angina to acute MI (see Table 15-5).1,2,3,6 An acute MI is generally described by patients as a “heart attack.” The following section discusses stable manifestations of CAD (stable angina) and acute manifestations described as an ACS (unstable angina and acute MI). Figure 15-1 is a concept map of ACS. Angina pectoris, or chest pain, caused by myocardial ischemia is not a separate disease, but rather a symptom of CAD. It is caused by a blockage or spasm of a coronary artery, leading to diminished myocardial blood supply. The lack of oxygen causes myocardial ischemia, which is felt as chest discomfort, pressure, or pain. Angina may occur anywhere in the chest, neck, arms, or back, but the most commonly described location is pain or pressure behind the sternum. The pain often radiates to the left arm but can also radiate down both arms and to the back, the shoulder, the jaw, or the neck (Fig. 15-2). Angina symptoms are not the same for all individuals, many patients may describe pressure or discomfort rather than pain, and presenting symptoms can be highly individualized, as described in Box 15-3. Patients and families must be taught that angina does not always present in the dramatic heart attack scenario, as often portrayed on television and in movies, in which the person clutches the throat or chest and exhibits extreme distress.2 Many women experience a variety of different symptoms before an acute MI and during the acute event, as shown in Box 15-4.32,33 The recognition and publicity about the fact that many women do not experience “crushing chest pain” is important to avoid women’s symptoms being trivialized by clinicians.37 It is important that women are made aware of angina symptom equivalents such as unexplained shortness of breath, breaking out in a cold sweat, or sudden fatigue, nausea, or lightheadedness.2 More women die every year in the United States of ACS compared with men, a fact that is largely unknown by health care providers.33,34 Box 15-4 Cardiovascular Symptoms Experienced by Women before Acute Myocardial Infarction From McSweeney JC, et al: Women’s early warning symptoms of acute myocardial infarction. Circulation. 2003;108(21):2619. Stable angina is predictable and caused by similar precipitating factors each time; typically, it is exercise induced. Patients become used to the pattern of this type of angina and may describe it as “my usual chest pain.” Pain control should be achieved within 5 minutes of rest and by taking sublingual nitroglycerin. Ischemia and chest pain occur when myocardial demand from exertion exceeds the fixed blood oxygen supply. Additional information on CAD and stable angina is provided in Box 15-5. Box 15-5 Evidence-Based Practice Coronary Artery Disease and Stable Angina • Diet low in salt and high in fiber, fruit, vegetables, and grains. • All dietary fat less than 30% of total calories; saturated fat less than 7%. • Limit glucose in diet (simple sugars). • Limit calories if overweight. • Start by walking more often and increase physical exercise from there. Refer to cardiac rehabilitation program. Strong evidence exists that the following diagnostic procedures help the patient with angina: • When a patient presents with chest pain, quickly obtaining a detailed history of symptoms, focused physical examination, and risk factor assessment can help determine whether the probability of CAD is low, intermediate, or high. • Initial laboratory tests include hemoglobin, fasting blood glucose, lipid panel, cardiac enzymes. • Obtain a baseline 12-lead ECG at rest, even if chest pain is not present. • Obtain a 12-lead ECG during an episode of chest pain. • Obtain a chest radiograph if symptoms of heart failure are present. • Obtain an exercise 12-lead ECG if the condition is stable and symptoms suggest CAD or if the condition is stable with complete LBBB or RBBB that makes the ECG difficult to interpret for ischemia. • Obtain cardiac echocardiography for patients with a systolic murmur suggestive of aortic stenosis. • Use cardiac echocardiography to determine extent of left ventricular (LV) hypertrophy or dysfunction. • Stress cardiac echocardiography is recommended for patients with greater than 1 mm of ST-segment depression at rest (stress may be by physical exercise or by pharmacologic stimulation). • Coronary angiography (typically as part of a cardiac catheterization procedure) is recommended for patients at high risk for adverse coronary events. Initial Pharmacologic and Lifestyle Treatment Recommendations • The goal of treatment is to eliminate chest pain. • The 10 most important elements of CAD and stable angina management can be remembered using the A to E mnemonic: A—Aspirin and antianginal medications: Prescribe daily, low-dose (75 to 325 mg) aspirin; oral nitrates; and sublingual nitroglycerin for episodes of angina. B—Beta-blockers and blood pressure: Use ACEI and beta-blockers to decrease blood pressure to less than 140/90 mm Hg if no other CAD risk factors are present and to less than 130/80 mm Hg if diabetes or kidney disease are present. C—Cholesterol and cigarettes: Obtain a fasting lipid profile. Recommend diet or lipid reduction medication therapy (statin) to lower LDL-C to less than 100 mg/dL (<70 mg/dL if achievable), increase HDL-C to more than 40 mg/dL for men or more than 50 mg/dL for women, and reduce triglycerides to less than 150 mg/dL. D—Diet and diabetes: Prescribe a low-fat, calorie-appropriate diet and provide nutritional consultation as needed to achieve a fasting blood glucose level of 70 to 100 mg/dL and HbA1c of less than 6.5%. E—Education and exercise: Provide education about risk factor modification and the CAD disease process; recommend daily exercise for 30 to 60 minutes (ideal) or at least seven times each week (minimum of 5 days per week). A BMI between 18.5 and 24.9 kg/m2 and waist less than 40 inches for men or less than 35 inches for women should be recommended. Treat depression, if present. HRT is not recommended as a treatment for symptoms of coronary heart disease. Influenza vaccination is recommended. Interventional and Surgical Recommendations for Stable High-Risk Patients Unstable angina is defined as a change in a previously established stable pattern of angina. It is part of the continuum of ACS. Unstable angina usually is more intense than stable angina, may awaken the person from sleep, or may necessitate more than nitrates for pain relief. A change in the level or frequency of symptoms requires immediate medical evaluation. Severe angina that persists for more than 5 minutes, worsens in intensity, and is not relieved by one nitroglycerin tablet is a medical emergency, and the patient or a family member must call 911 immediately.3 The 911 (Emergency Medical Services [EMS]) system is available to 90% of the population of the United States.3,35 Family and friends are discouraged from driving a person experiencing unstable angina to the hospital and instead are urged to call 911. Patients should be instructed never to drive themselves but to contact the EMS by calling 911. Unstable angina is an indication of atherosclerotic plaque instability. It can signal atherosclerotic plaque rupture and thrombus formation that can lead to MI. The patient who comes to the emergency department with recent-onset unstable angina but who has nonspecific or nonelevated ST-segment changes on the 12-lead electrocardiogram (ECG) may be admitted to the critical care unit to rule out MI. If the symptoms are typical of an MI, it is important to treat the patient according to the latest published guidelines because not all patients who experience an MI have ST-segment elevation on the 12-lead ECG.2,14,15 Variant angina, or Prinzmetal angina, is caused by a dynamic obstruction from intense vasoconstriction of a coronary artery.36 Spasm can occur with or without atherosclerotic lesions. Variant angina commonly occurs when the individual is at rest, and it is often cyclic, occurring at the same time every day. Smoking, alcohol use, and illegal stimulant drug (cocaine) use may precipitate spasm. A definitive diagnosis of variant angina is made during a cardiac catheterization study. Signs of spasm include ST-segment elevation and chest pain. Coronary artery spasm can occur with or without CAD. The prognosis is excellent when no significant coronary artery stenosis exists. Coronary artery spasm is treated with nitroglycerin or calcium channel blockers to vasodilate the coronary arteries. Silent ischemia describes a situation in which objective evidence of ischemia is observed on an ECG monitor but the person does not complain of anginal symptoms. Silent ischemia can occur in many clinical situations, as described in Box 15-6. One third of patients who are having a heart attack do not report chest pain as a symptom.2 Patients with diabetes are at particular risk for silent ischemia. Many patients who have had type 2 diabetes for more than 10 years have developed autonomic neuropathy, which decreases their ability to experience chest pain. Patients with diabetes may misinterpret angina-equivalent symptoms such as nausea, vomiting, and diaphoresis as signaling a disruption in glucose control rather than a sign of myocardial ischemia. Accurate assessment of chest pain symptoms is essential if unstable angina is to be recognized and treated effectively. Factors to consider when assessing chest pain are listed in Box 15-7. An important reason to ask questions about the chest pain is to differentiate between stable and unstable angina. The change from stable to unstable angina is potentially life threatening for the patient. If the ST segments are elevated or a newly documented left bundle branch block (LBBB) is seen on the 12-lead ECG, the patient should be treated for acute MI.2 However, if these classic ECG signs are missing and the chest pain continues, the current pharmacologic treatment of choice is aspirin (if the patient cannot tolerate aspirin, then a thienopyridine such as clopidogrel can be given). Patients with definite unstable angina or non–ST elevation myocardial infarction (UA or NSTEMI) should receive dual antiplatelet therapy on admission if an invasive strategy is imminent. A glycoprotein (GP) IIb/IIIa inhibitor is administered unless bivalirudin is chosen; and a loading dose of clopidogrel is given at least 6 hours prior to the procedure. Patients undergoing noninvasive treatment should receive aspirin and a thienopyridine (clopidgorel, prasugrel or ticragrelor) for 1 month and ideally up to 1 year. If symptoms persist, a diagnostic angiography is performed. A stress test should be performed on patients who are not undergoing invasive therapy for their UA/NSTEMI and if negative the GP IIb/IIIa inhibitor can be discontinued and unfractionated heparin (UFH) administered for 48 hours. Box 15-7 Factors to Consider When Assessing Chest Pain • Onset: Was it sudden or gradual? • Duration: Did pain last seconds or minutes? How soon after onset did the patient call for help? • Precipitating factors: Was the patient up and moving around? • Location: Was pain substernal? Was it located in same area as previous pain? • Radiation: Did pain radiate to the jaw, neck, arm, or shoulder? • Quality: Was pain similar to previous anginal pain? less painful or more painful? • Intensity: On a scale of 1 to 10, where would the patient rate the pain? • Relieving factors: What made the pain better—changing position, nitroglycerin, oxygen, the presence of the nurse? • Aggravating factors: Did things such as the environment, telephone calls, or waiting for help worsen the pain? • Associated symptoms: Was the pain accompanied by nausea, vomiting, diaphoresis, or dyspnea? • Emotional response: Was there an emotional response that intensified the pain—anxiety, fear, anger?
Cardiovascular Disorders
Coronary Artery Disease
Description and Etiology
Risk Factors for Coronary Artery Disease
Age, Gender, and Race
Family History
Hyperlipidemia
ATP III Classification of Total, Low-Density Lipoprotein (LDL), and High-Density Lipoprotein (HDL) Cholesterol and Triglycerides (mg/dL)*
Total Cholesterol
<200
200-239
Borderline-high
>240
High
LDL Cholesterol
<100
Optimal
100-129
Near or above optimal
130-159
Borderline high
160-189
High
>190
Very high
HDL Cholesterol
<40
Low
>60
High
Triglycerides
<150
Normal
150-199
Borderline high
200-499
High
>500
Very high
High-Density Lipoprotein Cholesterol.
Low-Density Lipoprotein Cholesterol.
Triglycerides.
Lipoprotein(a).
High-Fat Diet
Obesity
Physical Activity
Hypertension
CATEGORY
SYSTOLIC BP* (mm Hg)
DIASTOLIC BP* (mm Hg)
Normal (optimal)*
<120
<80
Prehypertension
120-139
80-89
Stage 1 hypertension
140-159
90-99
Stage 2 hypertension
≥160
≥100
Cigarette Smoking
Diabetes Mellitus
BLOOD GLUCOSE LEVEL
FASTING PLASMA GLUCOSE LEVEL* (mg/dL)
Normal
70-100
Prediabetes
100-125
Diabetes
126 or higher
Chronic Kidney Disease
Metabolic Syndrome
Women and Heart Disease
C-Reactive Protein
CATEGORY
HS-CRP LEVEL* (mg/L)
Low risk (normal)
<1
Moderate risk
1-3
High risk
>3
Coronary Artery Disease Risk Equivalents
Multifactorial Risk
Primary versus Secondary Prevention of Coronary Artery Disease
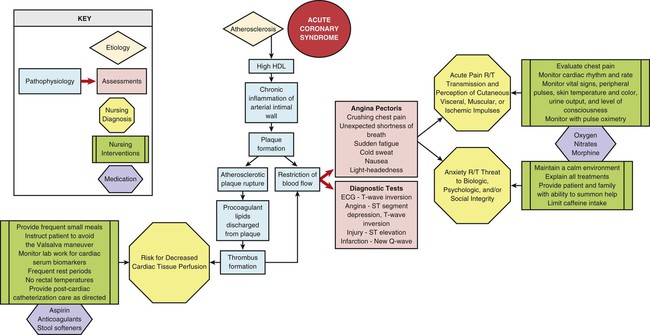
Pathophysiology of Coronary Artery Disease
Development of Atherosclerosis
Atherosclerotic Plaque Rupture
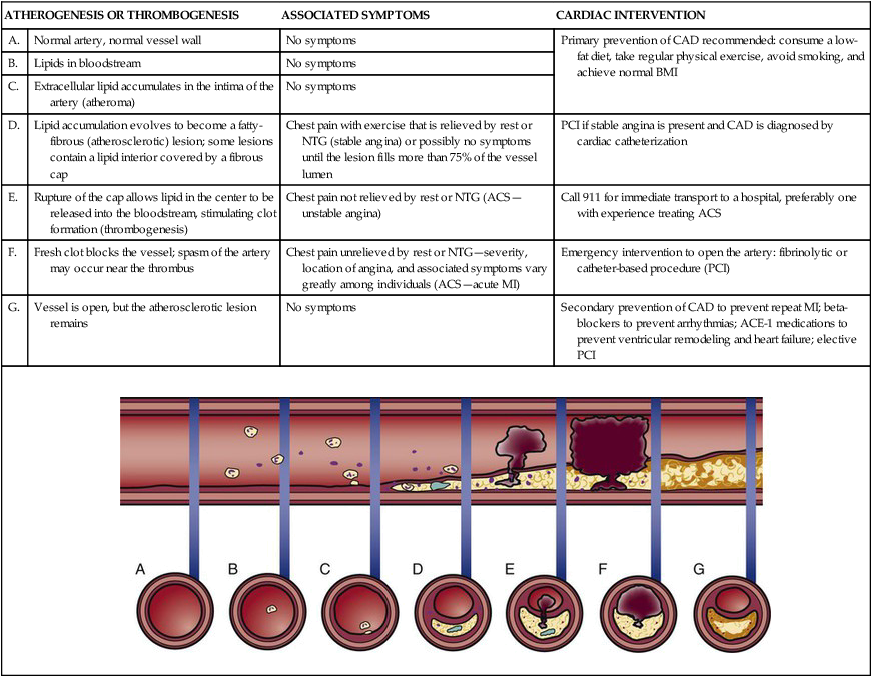
ATHEROGENESIS OR THROMBOGENESIS
ASSOCIATED SYMPTOMS
CARDIAC INTERVENTION
A.
Normal artery, normal vessel wall
No symptoms
Primary prevention of CAD recommended: consume a low-fat diet, take regular physical exercise, avoid smoking, and achieve normal BMI
B.
Lipids in bloodstream
No symptoms
C.
Extracellular lipid accumulates in the intima of the artery (atheroma)
No symptoms
D.
Lipid accumulation evolves to become a fatty-fibrous (atherosclerotic) lesion; some lesions contain a lipid interior covered by a fibrous cap
Chest pain with exercise that is relieved by rest or NTG (stable angina) or possibly no symptoms until the lesion fills more than 75% of the vessel lumen
PCI if stable angina is present and CAD is diagnosed by cardiac catheterization
E.
Rupture of the cap allows lipid in the center to be released into the bloodstream, stimulating clot formation (thrombogenesis)
Chest pain not relieved by rest or NTG (ACS—unstable angina)
Call 911 for immediate transport to a hospital, preferably one with experience treating ACS
F.
Fresh clot blocks the vessel; spasm of the artery may occur near the thrombus
Chest pain unrelieved by rest or NTG—severity, location of angina, and associated symptoms vary greatly among individuals (ACS—acute MI)
Emergency intervention to open the artery: fibrinolytic or catheter-based procedure (PCI)
G.
Vessel is open, but the atherosclerotic lesion remains
No symptoms
Secondary prevention of CAD to prevent repeat MI; beta-blockers to prevent arrhythmias; ACE-1 medications to prevent ventricular remodeling and heart failure; elective PCI
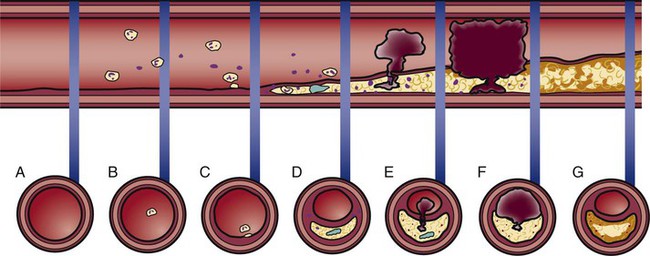
Plaque Regression
Acute Coronary Syndrome
Angina
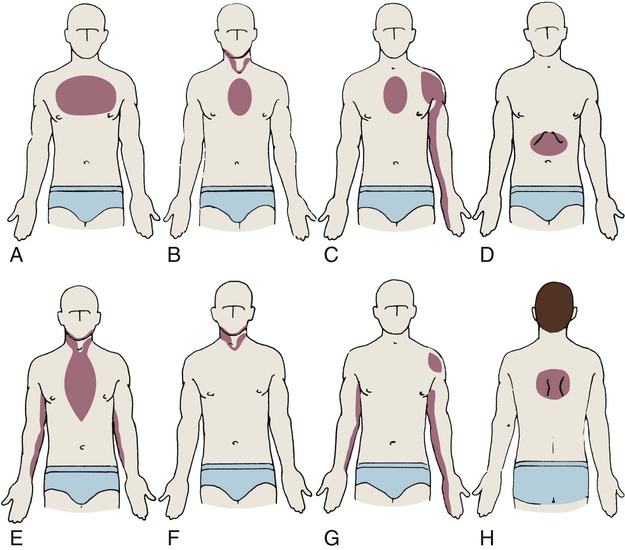
A, Upper part of chest; B, beneath sternum, radiating to neck and jaw; C, beneath sternum, radiating down left arm; D, epigastric; E, epigastric, radiating to neck, jaw, and arms; F, neck and jaw; G, left shoulder; H, intrascapular.
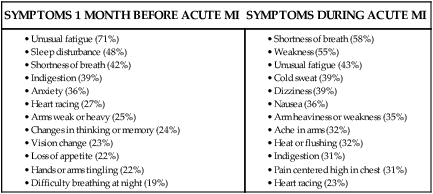
Women and Angina.
SYMPTOMS 1 MONTH BEFORE ACUTE MI
SYMPTOMS DURING ACUTE MI
Stable Angina.
 Strong evidence exists that the following lifestyle interventions help to prevent CAD:
Strong evidence exists that the following lifestyle interventions help to prevent CAD:
Unstable Angina.
Variant Angina.
Silent Ischemia.
Medical Management
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Cardiovascular Disorders
Get Clinical Tree app for offline access
