(bah-zih-LIX-ih-mab)
Simulect
Recombinant monoclonal antibody
Immunosuppressant
Usual dose
Basiliximab should only be administered once it has been determined that the patient will receive the graft and concomitant immunosuppression. Patients who have received a previous course of basiliximab should only be re-exposed to a subsequent course of therapy with extreme caution. Used concurrently with cyclosporine (Sandimmune) and corticosteroids.
Organ rejection prophylaxis in renal transplant:
2 doses of 20 mg each as an infusion. Administer the first dose within 2 hours before transplantation. Give the second dose 4 days after transplantation. Withhold the second dose if complications such as severe hypersensitivity reactions to basiliximab or graft loss occur.
Pediatric dose
Less than 35 kg:
2 doses of 10 mg each (discard remaining product after each dose).
35 kg or more:
2 doses of 20 mg each.
In all pediatric patients, administer the first dose within 2 hours before transplantation. Give the second dose 4 days after transplantation. See Actions and Maternal/Child. Withhold second dose if complications occur (e.g., hypersensitivity reactions or graft loss).
Dose adjustments
No dose adjustments indicated.
Dilution
Reconstitute each single 20-mg vial with 5 mL of SWFI. Reconstitute each 10-mg vial with 2.5 mL SWFI. Shake gently to dissolve powder. After reconstitution, may be given as an IV injection or may be further diluted to 50 mL with NS or D5W. When mixing, gently invert to avoid foaming; do not shake.
Storage:
Store unopened vials in the refrigerator (2° to 8° C [36° to 46° F]). Should be used within 4 hours of reconstitution. If necessary, may be refrigerated for up to 24 hours. Discard prepared solution after 24 hours.
Compatibility
Manufacturer states, “Other drug substances should not be added or infused simultaneously through the same IV line.” No incompatibilities observed with polyvinyl chloride bags and administration sets.
Rate of administration
May be given through a peripheral or central vein.
IV injection:
May be given as a bolus injection over 30 to 60 seconds. Incidence of N/V and local reaction including pain increased.
Infusion:
A single dose properly diluted over 20 to 30 minutes.
Actions
A chimeric (murine/human) monoclonal antibody produced by recombinant DNA technology. Functions as an immunosuppressant. Specifically binds to and blocks the interleukin-2 receptor alpha chain (IL-2Rα, also known as CD25 antigen), thereby inhibiting IL-2 driven proliferation of activated T-cells, which play a key role in organ rejection. Reduces/minimizes acute rejection. IL-2Rα is expressed selectively on activated, but not resting, T-cells. This selectivity prevents the profound generalized immunosuppression seen with other immunosuppressants used in organ transplantation and may decrease the risk of infection and development of lymphoproliferative disorders. Two 20-mg doses block the receptor for 4 to 6 weeks posttransplantation, the critical risk period for acute organ rejection. Has reduced the incidence of biopsy-confirmed acute rejections while minimizing side effects seen with other immunosuppressants. Clinical benefit demonstrated in a broad range of patients, regardless of age, gender, race, donor type, or history of diabetes mellitus as long as serum levels exceed 0.2 mg/mL (by ELISA). Mean half-life is 4 to 10.4 days. Half-life is increased (5.2 to 17.8 days) and distribution volume and clearance are decreased by about 50% in pediatric patients 2 to 11 years of age. Crosses the placental barrier. May be secreted in breast milk.
Indications and uses
Prophylaxis of acute organ rejection in patients receiving renal transplants. Used as part of an immunosuppressive regimen that includes cyclosporine and corticosteroids. Dosing regimen may also include either azathioprine or mycophenolate (CellCept IV).
Contraindications
Known hypersensitivity to basiliximab or any of its components (composite of human and murine antibodies).
Precautions
Usually administered by or under the direction of a physician experienced in immunosuppressive therapy and management of organ transplant patients. Adequate laboratory and supportive medical resources must be available. ■ Severe acute (onset within 24 hours) hypersensitivity reactions including anaphylaxis have been reported both with initial exposure and/or following re-exposure after several months. Emergency equipment and drugs for the treatment of severe hypersensitivity reactions must be readily available. Withhold the second dose of basiliximab if a hypersensitivity reaction occurs. ■ Readministration after an initial course of therapy has not been studied in humans, but other monoclonal antibodies have precipitated anaphylactoid reactions. ■ Potential for causing lymphoproliferative disorders, cytomegalovirus (CMV), and other opportunistic infections is unknown. ■ Use with caution in patients with infections or malignancies. ■ It is not known whether basiliximab use will have a long-term effect on the ability of the immune system to respond to antigens first encountered during induced immunosuppression. ■ Low titers of anti-idiotype antibodies and human antimurine antibodies (HAMA) to basiliximab have been detected in some patients during treatment; no adverse effects have been noted.
Monitor:
Obtain baseline CBC with differential and platelets and baseline renal and liver tests if not already completed for other immunosuppressant agents. ■ Monitor vital signs. ■ Observe closely for signs of infection (fever, sore throat, tiredness) or unusual bleeding or bruising. ■ Prophylactic antibiotics may be indicated pending results of C/S in a febrile immunosuppressed patient. ■ Symptoms of cytokine release syndrome (e.g., chills, fever, dyspnea, and malaise) have not been reported but may occur; observe carefully. ■ See Drug/Lab Interactions.
Patient education:
Report difficulty in breathing or swallowing, rapid heartbeat, rash, or itching immediately. ■ Report swelling of lower extremities and weakness. ■ Avoid pregnancy; nonhormonal birth control preferred. Women with childbearing potential should use effective contraception before beginning basiliximab therapy, during therapy, and for 2 months after completion. ■ See Appendix D, p. 1333.
Maternal/child:
Category B: use during pregnancy only if benefits justify the potential risk to the fetus. Avoid pregnancy; effective contraception required; see Patient Education. ■ Discontinue breast-feeding. ■ Has been used in pediatric patients from 2 to 15 years of age. No adequate or well-controlled studies completed. See differences in Actions. Most frequent side effects were fever and urinary infections.
Elderly:
Age-related dosing not required. Adverse events similar to younger adults. Use caution when giving immunosuppressive drugs to the elderly.
Drug/lab interactions
Has been administered concurrently with antilymphocyte globulin (ALG), antithymocyte globulin (ATG), azathioprine, corticosteroids, cyclosporine (Sandimmune), muromonab CD3 (Orthoclone), and mycophenolate (CellCept); no additional adverse reactions noted. ■ May increase or decrease numerous lab values, including serum calcium and potassium and fasting blood glucose.
Side effects
Basiliximab did not appear to alter the pattern, frequency, or severity of known side effects associated with the use of immunosuppressive drugs. Abdominal pain, anemia, constipation, diarrhea, edema, fever, headache, hyperkalemia, hypersensitivity reactions (including anaphylaxis, bronchospasm, capillary leak syndrome, cardiac failure, cytokine release syndrome, dyspnea, hypotension, pruritus, pulmonary edema, rash, respiratory failure, sneezing, tachycardia, urticaria, wheezing), hypertension, hypokalemia, insomnia, nausea, pain, peripheral edema, upper respiratory infections, urinary tract infections. Incidence of N/V and local reaction including pain increased with bolus injection. Severe hypersensitivity reactions (including anaphylaxis), capillary leak syndrome, and cytokine release syndrome have been reported. Incidence of infections, lymphomas, or other malignancies similar to placebo groups in studies.
Antidote
Notify physician of all side effects. Most will be treated symptomatically. Basiliximab may be discontinued or alternate immunosuppressive agents substituted. Discontinue immediately if anaphylaxis occurs, and treat with oxygen, epinephrine, corticosteroids, and/or antihistamines (e.g., diphenhydramine [Benadryl]). Resuscitate as necessary.
Belatacept 
(bel-AT-a-sept)
Nulojix
Immunosuppressant
pH 7.2 to 7.8
Usual dose
Administration of higher-than-recommended doses or more frequent dosing of belatacept is not recommended because of an increased risk of posttransplant lymphoproliferative disorder (PTLD), progressive multifocal leukoencephalopathy (PML), and serious CNS infections.
Premedication is not required. Base the total infusion dose on actual body weight at the time of transplantation.
| Dosing of Belatacept for Kidney Transplant Recipients* | |
| Dosing for Initial Phase | Dose |
| Day 1 (day of transplantation, before implantation) and Day 5 (approximately 96 hours after Day 1 dose) | 10 mg/kg |
| End of Week 2 and Week 4 after transplantation | 10 mg/kg |
| End of Week 8 and Week 12 after transplantation | 10 mg/kg |
| Dosing for Maintenance Phase | Dose |
| End of Week 16 after transplantation and every 4 weeks (plus or minus 3 days) thereafter | 5 mg/kg |

*The dose prescribed must be evenly divisible by 12.5 mg (evenly divisible increments are 0, 12.5, 25, 37.5, 50, 62.5, 75, 87.5, and 100). For example: At 10 mg/kg/dose, a patient weighing 64 kg would receive 640 mg. The closest doses evenly divisible by 12.5 below and above 640 mg are 637.5 mg and 650 mg. The nearest dose is 637.5 mg, and this would be the actual prescribed dose.
Regimen includes basiliximab (Simulect) induction, mycophenolate mofetil (MMF, CellCept), and corticosteroids.
Basiliximab:
20 mg IV on the day of transplantation and 4 days later.
Mycophenolate mofetil:
1 Gm twice daily as an initial dose. Adjust dose based on clinical signs of adverse events or efficacy failure.
Corticosteroid doses
should be consistent with those used in clinical trials. In clinical trials, the median corticosteroid doses were tapered to approximately 15 mg (10 to 20 mg) per day by the first 6 weeks and remained at approximately 10 mg (5 to 10 mg) per day for the first 6 months posttransplant; see Precautions. Actual corticosteroid dosing in clinical trials is summarized in the following chart.
| Actual Corticosteroid a Dosing in Studies 1 and 2 | ||
| Day of Dosing | Median (Q1-Q3) Daily Dose b, c | |
| Study 1 | Study 2 | |
| Week 1 | 31.7 mg (26.7 to 50 mg) | 30 mg (26.7 to 50 mg) |
| Week 2 | 25 mg (20 to 30 mg) | 25 mg (20 to 30 mg) |
| Week 4 | 20 mg (15 to 20 mg) | 20 mg (15 to 22.5 mg) |
| Week 6 | 15 mg (10 to 20 mg) | 16.7 mg (12.5 to 20 mg) |
| Month 6 | 10 mg (5 to 10 mg) | 10 mg (5 to 12.5 mg) |
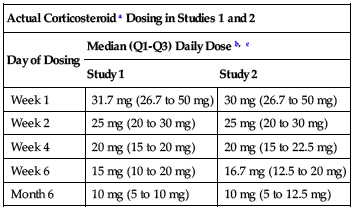
aCorticosteroid = Prednisone or prednisolone.
bProtocols allowed for flexibility in determining corticosteroid dose and rapidity of taper after Day 15. It is not possible to distinguish corticosteroid doses used to treat acute rejection versus doses used in a maintenance regimen.
cQ1 and Q3 are the 25th and 75th percentiles of daily corticosteroid doses, respectively.
Dose adjustments
Do not modify the dose during the course of therapy unless there is a change in body weight of greater than 10%. ■ Age, gender, race, renal function, hepatic function, diabetes, and concomitant dialysis do not affect the clearance of belatacept.
Dilution
Belatacept is for IV infusion only. Must be reconstituted/prepared using only the silicone-free disposable syringe provided with each vial. This syringe will be required for both reconstitution and the preparation of the final infusion. Maintain sterility. Any solution prepared with other than the provided silicone-free syringe must be discarded (contact the manufacturer to obtain an additional supply of silicone-free disposable syringes).
Each vial contains 250 mg of belatacept lyophilized powder. Calculate the number of vials needed to provide the total infusion dose. Reconstitute the contents of each vial with 10.5 mL of SWFI, NS, or D5W using the silicone-free disposable syringe and an 18- to 21-gauge needle. Direct the stream of diluent to the glass wall of the vial. To avoid foaming, rotate the vial and swirl gently until contents are completely dissolved. Avoid prolonged or vigorous agitation. Do not shake. Reconstituted solution yields 25 mg/mL and should be clear to slightly opalescent and colorless to pale yellow. Calculate the total volume of reconstituted solution required to provide the prescribed dose:
Volume of 25 mg/mL belatacept solution (in mL) = Prescribed dose (in mg) ÷ 25 mg/mL
The reconstituted solution must be further diluted in infusion fluid. If reconstituted with SWFI, dilute with either NS or D5W. If reconstituted with NS, further dilute with NS. If reconstituted with D5W, further dilute with D5W. From the appropriate-size infusion bag or bottle (typically an infusion volume of 100 mL is appropriate, but volumes from 50 to 250 mL may be used), withdraw a volume of infusion fluid equal to the total volume of reconstituted belatacept required to provide the prescribed dose. Using the same silicone-free disposable syringe used for reconstitution, withdraw the required amount of belatacept and inject it into the infusion bag or bottle. Gently rotate to ensure mixing. Concentration should range from 2 mg/mL to 10 mg/mL.
Filters:
Must be administered with a nonpyrogenic, low–protein binding, 0.2- to 1.2-micron filter.
Storage:
Refrigerate vials of lyophilized powder at 2° to 8° C (36° to 46° F) in carton to protect from light. The reconstituted solution should be further diluted in infusion fluid immediately. The infusion must be completed within 24 hours. Infusion solution can be refrigerated protected from light for up to 24 hours. A maximum of 4 hours of those 24 hours can be at RT 20° to 25° C (68° to 77° F) and room light. Discard any unused solution remaining in vials.
Compatibility
Manufacturer states, “Must be reconstituted/prepared using only the silicone-free disposable syringe provided with each vial. Any solution prepared with other than the provided silicone-free syringe must be discarded” and “Infuse in a separate line; should not be infused concomitantly in the same IV line with other agents.”
Rate of administration
A single dose evenly distributed over 30 minutes.
Actions
A selective T-cell costimulation blocker. Produced by recombinant DNA technology. Binds to CD80 and CD86 on antigen-presenting cells, thereby blocking CD28-mediated costimulation of T-lymphocytes. Belatacept binds to CD80 and CD86 more readily than abatacept (the parent molecule from which it is derived). Belatacept-mediated co-stimulation blockade results in the inhibition of cytokine production by T-cells required for antigen-specific antibody production by B-cells and inhibits T-lymphocyte proliferation. Activated T-lymphocytes are the predominant mediators of immunologic rejection. Half-life ranges from 6.1 to 15.1 days. In clinical trials, trough concentrations were consistently maintained from 6 months up to 3 years posttransplant. Increasing body weight may result in a trend toward higher clearance.
Indications and uses
Prophylaxis of organ rejection in adult patients receiving a kidney transplant. Used in combination with basiliximab (Simulect) induction, mycophenolate mofetil (CellCept), and corticosteroids.
Limitation of use:
Use only in patients who are Epstein-Barr virus (EBV) seropositive. ■ Use for prophylaxis of organ rejection in transplanted organs other than the kidney not established. Not recommended for use in liver transplant patients; risk of graft loss and death is increased.
Contraindications
Transplant recipients who are Epstein-Barr virus (EBV) seronegative or who have unknown EBV serostatus because of risk of posttransplant lymphoproliferative disorder (PTLD) predominantly involving the CNS.
Precautions
For IV use only. ■ Usually administered by or under the direction of a physician experienced in immunosuppressive therapy and management of organ transplant patients. Adequate laboratory and supportive medical resources must be available. ■ Increased risk for developing posttransplant lymphoproliferative disorder (PTLD), predominantly involving the CNS, compared with patients on a cyclosporine-based regimen. Recipients without immunity to Epstein-Barr virus (EBV) are at a particularly increased risk; therefore use in EBV-seropositive patients only. Do not use belatacept in transplant recipients who are EBV-seronegative or who have unknown EBV serostatus. ■ Other known risk factors for PTLD include cytomegalovirus (CMV) infection and T-cell–depleting therapy. Use T-cell–depleting therapies to treat acute rejection with caution. CMV prophylaxis is recommended for at least 3 months after transplantation. Patients who are EBV seropositive and CMV seronegative may be at increased risk for PTLD compared with patients who are EBV seropositive and CMV seropositive. ■ Minimization of the corticosteroid dose to 5 mg/day between Day 3 and Week 6 posttransplantation has been associated with an increased rate and grade of acute rejection, particularly Grade III rejection. Graft loss occurred in some patients. Corticosteroid use should be consistent with clinical trial experience; see Usual Dose. ■ Increased susceptibility to infection and possible development of malignancies may result from immunosuppression. Increased risk of developing other malignancies, including malignancies of the skin, appears related to intensity and duration of use. Avoid prolonged exposure to UV light and sunlight. Risk of developing bacterial, viral (e.g., CMV and herpes), fungal, and protozoal infections, including opportunistic infections such as tuberculosis (TB) or polyoma virus–associated nephropathy (PVAN), is increased and may lead to serious (including fatal) outcomes. Prophylaxis for Pneumocystis jiroveci is recommended after transplantation. ■ PML (a rapidly progressive and fatal opportunistic infection of the CNS that is caused by the JC virus) has been reported. ■ Infusion-related reactions have occurred within 1 hour of infusion; however, no serious reactions or anaphylaxis have been reported. ■ Use in liver transplant patients is not recommended; see Limitation of Use. ■ Anti-belatacept antibody development was not associated with an altered clearance of belatacept. The clinical impact of anti-belatacept antibodies has not been determined. ■ Do not administer live virus vaccines; see Drug/Lab Interactions.
Monitor:
Ascertain EBV serology before starting therapy with belatacept. ■ Monitor for new or worsening neurologic, cognitive, or behavioral signs and symptoms. May indicate PTLD or PML. ■ Evaluate patients for latent tuberculosis (TB). Patients testing positive in TB screening should be treated with a standard TB regimen before initiating belatacept therapy. ■ Monitor for S/S of infection; see Antidote. ■ Monitor renal function closely and consider PVAN if renal function is deteriorating. ■ New-onset diabetes, dyslipidemia, and hypertension may occur; monitor blood sugar, lipid panel, and BP. ■ See Precautions.
Patient education:
Read manufacturer’s patient information sheet before each infusion. ■ Risk of other malignancies, especially skin cancer, is increased. Limit exposure to sunlight and UV light. Wear protective clothing and use a sunscreen with a high protection factor. ■ Promptly report confusion, thinking problems, and loss of memory; decreased strength or weakness on one side of the body; and changes in mood or behavior, walking or talking, and vision. ■ Promptly report S/S of infection (e.g., fever, malaise). Adherence to prescribed antimicrobial prophylaxis is imperative. ■ May cause fetal harm. Pregnancy Registry available to monitor maternal-fetal outcomes.
Maternal/child:
Category C: may cause fetal harm. Use only if potential benefit to mother outweighs potential risk to fetus. ■ Discontinue breast-feeding. ■ Safety and effectiveness for use in pediatric patients less than 18 years of age not established.
Elderly:
Safety or effectiveness similar to that seen in younger adults.
Drug/lab interactions
A change of mycophenolic acid (MPA) exposure may occur with a crossover from cyclosporine to belatacept or from belatacept to cyclosporine in patients concomitantly receiving MMF. Cyclosporine decreases MPA exposure by preventing enterohepatic recirculation of MPA, whereas belatacept does not. A higher MMF dosage may be needed after switching from belatacept to cyclosporine because cyclosporine may result in lower MPA concentrations and increase the risk of graft rejection. A lower MMF dosage may be needed after switching from cyclosporine to belatacept because belatacept may result in higher MPA concentrations and increase the risk for adverse reactions related to MPA. ■ No dosage adjustments are needed for drugs metabolized via CYP1A2 (e.g., caffeine), CYP2C9 (e.g., losartan [Cozaar]), CYP2D6 (e.g., dextromethorphan), CYP3A (e.g., midazolam [Versed]), and CYP2C19 (e.g., omeprazole [Prilosec]). ■ Avoid the use of live vaccines during treatment with belatacept, including but not limited to intranasal influenza, measles, mumps, rubella, oral polio, BCG, yellow fever, varicella, and Ty21a typhoid vaccines.
Side effects
Anemia, constipation, cough, diarrhea, fever, graft dysfunction, headache, hyperkalemia, hypertension, hypokalemia, leukopenia, nausea, peripheral edema, urinary tract infection, and vomiting are most common. Posttransplant lymphoproliferative disorder (PTLD), predominantly CNS PTLD; other malignancies; and serious infections, including JC virus–associated progressive multifocal leukoencephalopathy (PML) and polyoma virus-associated nephropathy (PVAN), are the most serious potential side effects and may be life threatening. Abdominal pain, acne, anxiety, arthralgia, back pain, bronchitis, cytomegalovirus (CMV) and herpes infections, dyslipidemia, dysuria, hematuria, hypercholesterolemia, hyperglycemia, hyperuricemia, hypocalcemia, hypomagnesemia, hypophosphatemia, hypotension, influenza, insomnia, nasopharyngitis, proteinuria, renal tubular necrosis, tremor, tuberculosis, and upper respiratory infections may occur.
Antidote
Notify physician of all side effects. Most will be treated symptomatically. If PML is suspected, confirmation of the diagnosis by consultation with a neurologist, brain imaging, CSF testing for JC viral DNA, and/or brain biopsy is indicated. If PML is confirmed, consider reducing or discontinuing immunosuppression, taking into account the risk to the allograft. Therapy may need to be interrupted in patients who develop infections. Treat infusion reactions as indicated. Resuscitate as necessary.
Belimumab
(be-LIM-ue-mab)
Benlysta
Monoclonal antibody
pH 6.5
Usual dose
Premedication:
Consider premedication (e.g., antihistamines [e.g., diphenhydramine (Benadryl)], H2 antagonists [e.g., ranitidine (Zantac)], and/or corticosteroids [e.g., dexamethasone (Decadron)]) to help prevent or minimize hypersensitivity/infusion reactions.
Belimumab:
10 mg/kg as an infusion at 2-week intervals for the first 3 doses. Administer at 4-week intervals thereafter.
Dilution
Available in two strengths (120 mg or 400 mg). Allow vial to reach RT before reconstitution (approximately 10 to 15 minutes). Reconstitute the 120-mg vial with 1.5 mL SWFI and the 400-mg vial with 4.8 mL SWFI. Concentration for both will equal 80 mg/mL. Direct the SWFI toward the side of the vial to minimize foaming. Gently swirl for 60 seconds. Allow to sit at RT and gently swirl for 60 seconds every 5 minutes until completely dissolved. Do not shake. Reconstitution may take up to 30 minutes. Protect from sunlight. Solution should be opalescent and colorless to pale yellow. Small air bubbles are expected and are acceptable. If reconstituted with a mechanical swirler, do not exceed 500 rpm and/or a duration of 30 minutes. Desired dose of the reconstituted solution must be further diluted to 250 mL with NS by withdrawing and discarding a volume equal to the desired dose from a 250-mL infusion bag or bottle of NS. Add the desired dose of belimumab to the infusion bag or bottle and invert to mix the solution; see Storage.
Filters:
No data available from manufacturer.
Storage:
Before use, refrigerate vials (2° to 8° C [36° to 46° F]) in original carton, protected from light. Do not freeze. Avoid exposure to heat. Do not use beyond expiration date. Reconstituted solution, if not used immediately, should be refrigerated protected from direct sunlight. Solution diluted in NS may be refrigerated or kept at RT. Total time from reconstitution to completion of the infusion should not exceed 8 hours. Discard any unused product.
Compatibility
Manufacturer states, “Incompatible with dextrose solutions,” “No incompatibilities with polyvinylchloride or polyolefin bags observed,” and “Should not be infused concomitantly in the same IV line with other agents.”
Rate of administration
A single dose, properly diluted, as an infusion equally distributed over 1 hour. Slow or interrupt infusion rate if an infusion reaction develops.
Actions
A recombinant, DNA-derived, humanized monoclonal antibody. It is a B-lymphocyte stimulator (BLyS)–specific inhibitor that blocks the binding of soluble BLyS (a B-cell survival factor) to its receptors on B-cells. Does not bind with B-cells directly. However, by binding BLyS, it inhibits the survival of B-cells (including auto-reactive B-cells) and reduces the differentiation of B-cells into immunoglobulin-producing plasma cells. Significantly reduces circulating CD19+, CD20+, naïve, and activated B-cells, plasmacytoid cells, and the SLE B-cell subset. Reductions in IgG and anti-dsDNA and increases in complement (C3 and C4) were observed as early as Week 8 and sustained through Week 52. Terminal half-life is 19.4 days.
Indications and uses
Treatment of adult patients with active, autoantibody-positive, systemic lupus erythematosus (SLE) who are receiving standard therapy.
Limitation of use:
Not recommended in patients with severe active lupus nephritis or severe active central nervous system lupus; has not been studied in these situations.
Contraindications
Patients who have experienced an anaphylactic hypersensitivity reaction with belimumab.
Precautions
For IV infusion only. ■ Administered under the direction of a physician knowledgeable in its use in a facility with adequate diagnostic and treatment facilities to monitor the patient and respond to any medical emergency. ■ Serious hypersensitivity/infusion reactions, including anaphylaxis and death, have occurred. Acute hypersensitivity reactions usually occur within hours of the infusion. Nonacute reactions, including rash, nausea, fatigue, myalgia, headache, and facial edema, may occur up to a week following the infusion. Patients with a history of multiple drug allergies or significant hypersensitivity may be at increased risk. Administration of premedications (which can mask or mitigate a reaction) and the overlap in S/S may make it difficult to distinguish between a hypersensitivity reaction and an infusion reaction. ■ Serious and sometimes fatal infections have been reported. Use with caution in patients with chronic infections. Therapy should not be started if a patient is receiving any therapy for a chronic infection. ■ Cases of JC virus–associated progressive multifocal leukoencephalopathy (PML) resulting in neurologic deficits, including fatal cases, have been reported. Risk factors for PML include treatment with immunosuppressant therapies and impairment of immune function. ■ Psychiatric events, including depression, insomnia, and anxiety, have been reported. Most patients had a history of depression or other serious psychiatric disorders and were receiving psychoactive medications. ■ More deaths were reported with belimumab than with placebo during the controlled period of clinical trials. Etiologies included cardiovascular disease, infection, and suicide. No single cause of death predominated. ■ The impact of treatment with belimumab on the development of malignancies is not known. ■ Anti-belimumab antibodies developed in a small percentage of patients. Clinical relevance is unknown. ■ Response rates were lower in black/African-American patients; use with caution. ■ Use in patients with impaired renal or hepatic function has not been studied.
Monitor:
Obtain a baseline CBC, including a differential and platelet count. Monitor as indicated. ■ Monitor for hypersensitivity reactions carefully during and for an appropriate period of time after administration; delayed onset of severe hypersensitivity reactions has been reported. S/S reported include anaphylaxis, angioedema, dyspnea, hypotension, pruritus, rash, and urticaria. ■ On the day of the infusion, monitor for S/S of an infusion reaction (e.g., bradycardia, headache, hypotension, myalgia, nausea, rash, urticaria). ■ Monitor for S/S of PML, such as new-onset or deteriorating neurologic signs. Consultation with a neurologist is recommended. ■ Consider interrupting therapy if a new infection develops during treatment with belimumab.
Patient education:
Women of childbearing potential should use effective contraceptive methods during treatment and for a minimum of 4 months after the final treatment. ■ Manufacturer has established a pregnancy registry, and patients who are or who become pregnant are encouraged to register. ■ Tell your health care provider if you are allergic to any medications. ■ Promptly report difficulty breathing; dizziness or fainting; headache; itching; nausea; skin rash, redness, or swelling; swelling of the face, lips, mouth, tongue, or throat; may indicate a hypersensitivity/infusion reaction. ■ Promptly report bloody diarrhea, chest discomfort or pain, chills, cold sweats, coughing up mucus, dizziness, fever, nausea, new or worsening depression or suicidal thoughts, pain or burning with urination, unusual changes in behavior or mood. ■ Promptly report new or worsening neurologic symptoms (e.g., confusion, difficulty talking or walking, loss of balance, memory loss, or vision problems).
Maternal/child:
Category C: use during pregnancy only if the potential benefit justifies the potential risk to the fetus. ■ Discontinue breast-feeding. ■ Safety and effectiveness for use in pediatric patients not established.
Elderly:
Numbers in clinical studies are insufficient to determine if the elderly respond differently than younger subjects; use with caution.
Drug/lab interactions
Formal drug interaction studies have not been performed. ■ Has not been studied in combination with other biologics, including B-cell–targeted therapies or IV cyclophosphamide; concurrent use is not recommended. ■ Do not administer live virus vaccines for 30 days before or concurrently with belimumab. ■ Has been administered concomitantly with corticosteroids, antimalarials, immunomodulatory and immunosuppressive agents (including azathioprine, methotrexate, and mycophenolate), angiotensin-pathway antihypertensives, HMG-CoA reductase inhibitors (statins), and NSAIDs without meaningful clinical effect on belimumab pharmacokinetics.
Side effects
Bronchitis, depression, diarrhea, fever, insomnia, nasopharyngitis, migraine, nausea, pain in extremities, and pharyngitis were most commonly reported. The most common serious infections included bronchitis, cellulitis, pneumonia, and urinary tract infections. Anxiety, cystitis, depression, hypersensitivity/infusion reactions, influenza, leukopenia, sinusitis, and viral gastroenteritis also were reported.
Post-marketing:
Fatal anaphylaxis.
Antidote
Notify physician of all side effects. Treatment of most side effects will be supportive. Consider interrupting therapy if a new infection develops during treatment with belimumab. If PML is confirmed, consider discontinuing immunosuppressant therapy, including belimumab. Slow or interrupt infusion rate if an infusion reaction develops. Infusion reactions may be treated with acetaminophen, antiemetics (e.g., ondansetron [Zofran]), antihistamines (e.g., diphenhydramine [Benadryl]), H2 antagonists (e.g., ranitidine [Zantac]), or corticosteroids (e.g., hydrocortisone sodium succinate [Solu-Cortef]) as indicated. Discontinue if a serious hypersensitivity reaction occurs. Treat hypersensitivity reactions as indicated; may require epinephrine, airway management, oxygen, IV fluids, antihistamines (e.g., diphenhydramine [Benadryl]), corticosteroids (e.g., hydrocortisone sodium succinate [Solu-Cortef]), and pressor amines (e.g., dopamine).
Belinostat
(be-LIN-oh-stat)
Beleodaq
Antineoplastic
Usual dose
1,000 mg/M2 administered over 30 minutes by IV infusion once each day on Days 1 through 5 of a 21-day cycle. Cycles can be repeated every 21 days until disease progression or unacceptable toxicity.
Dose adjustments
Before the start of each cycle and before resuming treatment following toxicity, the absolute neutrophil count (ANC) should be greater than or equal to 1 × 109/L (1,000/mm3), and the platelet count should be greater than or equal to 50 × 109/L (50,000/mm3). ■ Other toxicities must be NCI-CTCAE Grade 2 or less before retreatment. ■ Reduce the starting dose of belinostat to 750 mg/M2 in patients known to be homozygous for the UGTIA*28 allele. ■ See the following chart for dose modifications for hematologic and nonhematologic toxicities. Base the dose adjustments for thrombocytopenia and neutropenia on platelet and absolute neutrophil nadir (lowest value) counts in the preceding cycle of therapy.
| Dose Modifications for Hematologic and Nonhematologic Toxicities | |
| Hematologic Toxicities | Dose Modification |
| Platelet count ≥25 × 109/L and nadir ANC ≥0.5 × 109/L | No change |
| Nadir ANC <0.5 × 109/L (any platelet count) | Decrease dose by 25% (750 mg/M2) |
| Platelet count <25 × 109/L (any nadir ANC) | Decrease dose by 25% (750 mg/M2) |
| Nonhematologic Toxicities | Dose Modification |
| Any CTCAE Grade 3 or 4 adverse reaction* | Decrease dose by 25% (750 mg/M2) |
| Recurrence of CTCAE Grade 3 or 4 adverse reaction after two dose reductions | Discontinue Beleodaq |
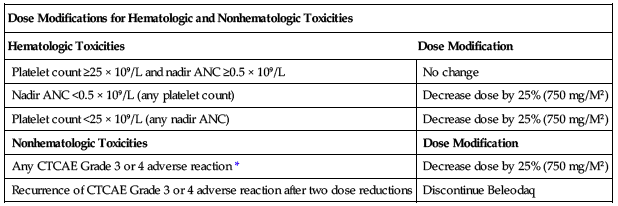
*For nausea, vomiting, and diarrhea, dose modify only if the duration is greater than 7 days with supportive management.
Dilution
Specific techniques required; see Precautions. Available in single-use vials containing lyophilized powder equivalent to 500 mg belinostat. Reconstitute each vial with 9 mL of SWI (concentration is 50 mg/mL). Swirl contents until no visible particles remain. Withdraw the required dose and transfer to an infusion bag containing 250 mL of NS. Do not use if cloudiness or particulates are observed.
Filters:
Use of an infusion set with a 0.22-micron in-line filter is required for administration.
Storage:
Store in original packaging at CRT until use. Reconstituted vials may be stored for up to 12 hours at (15° to 25° C (59° to 77° F). Fully diluted solution may be stored at 15° to 25° C (59° to 77° F) for up to 36 hours, including infusion time.
Compatibility
Specific information not available.
Rate of administration
A single dose as an IV infusion equally distributed over 30 minutes. Infusion time may be extended to 45 minutes for infusion site pain or other symptoms potentially attributable to the infusion.
Actions
A histone deacetylase (HDAC) inhibitor. HDACs catalyze the removal of acetyl groups from the lysine residues of histones and some nonhistone proteins. In vitro, belinostat caused the accumulation of acetylated histones and other proteins, inducing cell-cycle arrest and/or apoptosis of some transformed cells. It shows preferential cytotoxicity toward tumor cells compared with normal cells. Highly bound to protein. Primarily metabolized by UGT1A1. Also undergoes some metabolism by selected cytochrome P450 isoenzymes. Elimination half-life is 1.1 hours. Excreted in urine.
Indications and uses
Treatment of patients with relapsed or refractory peripheral T-cell lymphoma. Approval is based on tumor response rate and duration of response. An improvement in survival or disease-related symptoms has not been established.
Contraindications
Manufacturer states, “None.”
Precautions
Follow guidelines for handling cytotoxic agents. See Appendix A, p. 1331. ■ Usually administered by or under the direction of a physician specialist with adequate diagnostic and treatment facilities to monitor the patient and respond to any medical emergency. ■ Can cause thrombocytopenia, leukopenia (neutropenia and lymphopenia), and/or anemia. See Monitor and Dose Adjustments. ■ Serious and sometimes fatal infections, including pneumonia and sepsis, have occurred. Do not administer belinostat to patients with an active infection. Patients with a history of extensive or intensive chemotherapy may be at higher risk for life-threatening infections. ■ Can cause fatal hepatotoxicity and liver function test abnormalities. Interrupt or adjust dose until recovery, or permanently discontinue belinostat based on the severity of the hepatic toxicity. ■ Tumor lysis syndrome (TLS) has occurred. May cause acute renal failure requiring dialysis and can be fatal. ■ Gastrointestinal toxicity has been reported. ■ Use caution in patients with hepatic impairment. Patients with moderate and severe hepatic impairment (total bilirubin greater than 1.5 × ULN) were excluded from clinical trials. ■ See Monitor.
Monitor:
Obtain baseline CBC, including platelets, and monitor weekly. ■ Obtain baseline serum chemistry tests, including renal and hepatic function, before the start of the first dose of each cycle. ■ Observe closely for signs of infection. Prophylactic antibiotics may be indicated pending results of C/S in a febrile neutropenic patient. ■ Monitor for S/S of TLS; early signs are flank pain and hematuria. May further develop to a rapid reduction in tumor volume, renal insufficiency, hyperkalemia, hypocalcemia, hyperuricemia, or hyperphosphatemia. Monitoring of serum electrolytes, uric acid, and renal function indicated. ■ Prevention and treatment of hyperuricemia due to TLS may be accomplished with adequate hydration and, if necessary, allopurinol (Aloprim) and alkalinization of urine. ■ Monitor for thrombocytopenia (platelet count less than 50,000 mm3). Initiate precautions to prevent excessive bleeding (e.g., inspect IV sites, skin, and mucous membranes; use extreme care during invasive procedures; test urine, emesis, stool, and secretions for occult blood). ■ Nausea, vomiting, and diarrhea may require the use of antiemetic and/or antidiarrheal medications. Use prophylactically to increase patient comfort.
Patient education:
Review manufacturer’s patient information leaflet. ■ Avoid pregnancy. Nonhormonal birth control recommended. ■ Promptly report nausea, vomiting, and diarrhea; antiemetics and/or antidiarrheals may be indicated. ■ Promptly report symptoms of infection (e.g., fever). ■ Report bloody stools, bruising, fatigue. ■ Monitoring of liver function is imperative. ■ See Appendix D, p. 1333.
Maternal/child:
Category D: avoid pregnancy. A genotoxic drug that targets rapidly dividing cells; may cause teratogenicity and/or embryo-fetal death. May impair male fertility. ■ Discontinue breast-feeding. ■ Safety and effectiveness for use in pediatric patients not established.
Elderly:
Patients 65 years of age and older had a higher response rate to belinostat than did younger adults. ■ No clinically meaningful differences in serious side effects were observed in patients based on age.
Drug/lab interactions
Avoid concomitant administration of belinostat with strong inhibitors of UGT1A1 (e.g., atazanavir [Reyataz], indinavir [Crixivan], ketoconazole [Nizoral]). ■ Belinostat did not increase the AUC or Cmax of warfarin (Coumadin). Dose adjustment of warfarin is not required when coadministered with belinostat. ■ Likely a glycoprotein (P-gp) substrate but not likely to inhibit P-gp.
Side effects
The most common side effects reported are anemia, fatigue, fever, nausea and vomiting. Serious side effects reported include hematologic toxicity (anemia, lymphopenia, neutropenia, thrombocytopenia), hepatotoxicity and liver function abnormalities, increased SCr, infections (pneumonia, sepsis), multiorgan failure, and tumor lysis syndrome. Anemia, fatigue, febrile neutropenia, and multiorgan failure were reported as the reason for discontinuation of treatment. Other side effects reported include abdominal pain, chills, constipation, cough, decreased appetite, diarrhea, dizziness, dyspnea, headache, hypokalemia, hypotension, increased blood lactate dehydrogenase, infusion site pain, peripheral edema, phlebitis, prolonged QT, pruritus, and rash. Treatment-related deaths have occurred.
Antidote
Notify physician of all side effects. Minor side effects may be treated symptomatically. Supportive therapy as indicated will help sustain the patient in toxicity. Interrupt or adjust dose until recovery, or permanently discontinue belinostat based on the severity of the hematologic or hepatic toxicity. Hematologic toxicity may require dose adjustment. Neutropenia may be treated with filgrastim (Neupogen, Zarxio). Severe thrombocytopenia or anemia may require transfusion. Should a hypersensitivity reaction occur, treat as indicated.
Bendamustine hydrochloride
(ben-deh-MUS-teen)
Bendeka, Treanda
Antineoplastic
Alkylating agent
pH 2.5 to 3.5
Usual dose
Premedication:
Premedication with antihistamines (e.g., diphenhydramine [Benadryl]), antipyretics (e.g., acetaminophen [Tylenol]), and corticosteroids may be indicated; see Monitor and Antidote.
Chronic lymphocytic leukemia (CLL):
100 mg/M2 as an IV infusion on Days 1 and 2 of a 28-day cycle. May be repeated for up to 6 cycles.
Non-Hodgkin’s lymphoma (NHL):
120 mg/M2 as an IV infusion on Days 1 and 2 of a 21-day cycle. May be repeated for up to 8 cycles.
Dose adjustments
Chronic lymphocytic leukemia (CLL) and non-Hodgkin’s lymphoma (NHL):
Delay treatment for Grade 4 hematologic toxicity or clinically significant nonhematologic toxicity equal to or greater than Grade 2. In addition, dose reduction may be indicated; see specific indication. Reinitiate treatment, if indicated, when nonhematologic toxicity has recovered to equal to or less than Grade 1 and/or the ANC has recovered to equal to or greater than 1,000 cells/mm3 and platelets have recovered to equal to or greater than 75,000 cells/mm3. ■ No dose adjustment indicated based on age or gender; see Precautions.
Chronic lymphocytic leukemia (CLL):
Reduce dose to 50 mg/M2 on Days 1 and 2 of each cycle for Grade 3 or greater hematologic toxicity. If Grade 3 or greater toxicity recurs, reduce dose to 25 mg/M2 on Days 1 and 2 of each cycle. ■ Reduce dose to 50 mg/M2 on Days 1 and 2 of each cycle for clinically significant Grade 3 or greater nonhematologic toxicity. ■ Dose re-escalation in subsequent cycles may be considered by the treating physician.
Non-Hodgkin’s lymphoma (NHL):
Reduce dose to 90 mg/M2 on Days 1 and 2 of each cycle for Grade 4 hematologic toxicity. If Grade 4 toxicity recurs, reduce the dose to 60 mg/M2 on Days 1 and 2 of each cycle. ■ Reduce dose to 90 mg/M2 on Days 1 and 2 of each cycle for Grade 3 or greater nonhematologic toxicity. If Grade 3 or greater toxicity recurs, reduce the dose to 60 mg/M2 on Days 1 and 2 of each cycle.
Dilution
Specific techniques required; see precautions.
Treanda is available in two formulations: a solution (Treanda Injection) and a lyophilized powder (Treanda for Injection). Formulations have different concentrations. Do not mix or combine the two formulations.
Treanda Injection is available as a solution in 45 mg/0.5 mL and 180 mg/2 mL single-use vials. Do not use with devices containing polycarbonate or acrylonitrile-butadiene-styrene (ABS), including closed-system transfer devices (CSTDs), adaptors, and syringes when preparing the infusion bag. Must be diluted in a biosafety cabinet or a containment isolator. Aseptically withdraw the volume needed for the required dose and transfer to a 500-mL infusion bag of NS or D2.5/1/2NS using a polypropylene syringe with a metal needle and polypropylene hub. (Polypropylene syringes are translucent in appearance.) Final concentration of infusion solution should be 0.2 to 0.7 mg/mL. Mix thoroughly; solution should be clear and colorless to slightly yellow; see Compatibility. After dilution of Treanda Injection in the infusion bag, devices that contain ABS or polycarbonate, including infusion sets, may be used.
Treanda for Injection is available as a lyophilized powder in 25- and 100-mg single-use vials. If a CSTD or adaptor that contains polycarbonate or ABS is to be used as supplemental protection during preparation, use only this formulation (Treanda for Injection). Reconstitute each 25-mg vial with 5 mL SWFI and each 100-mg vial with 20 mL SWFI; concentration is 5 mg/mL. Shake well. Should completely dissolve in 5 minutes. Within 30 minutes of reconstitution, withdraw the desired dose from the vial(s) and further dilute in 500 mL of NS or D2.5/1/2NS. Final concentration should be 0.2 to 0.6 mg/mL. Mix thoroughly; solution should be clear and colorless to slightly yellow.
Bendeka is available as a ready-to-dilute solution in 100 mg/4 mL multiple-dose vials. Withdraw the volume needed for the required dose and immediately transfer into a 50-mL infusion bag of NS, D5W, or D2.5/1/2NS. The final concentration should be 1.85 to 5.6 mg/mL. The final admixture should be a clear and colorless to yellow solution.
Generic bendamustine hydrochloride as a lyophilized powder has recently been approved. Prescribing information was not available at the time of this printing. Read labeling carefully. Product formulation, dilution, storage, and precautions around administration may differ among the various products.
Filters:
Specific information not available.
Storage:
All formulations:
Retain in original package to protect from light. Bendeka must be stored in the refrigerator at 2° to 8° C (36° to 46° F). Solution diluted in NS or D2.5/1/2NS is stable for 6 hours at RT (15° to 30° C [59° to 86° F]) and room light or for 24 hours refrigerated at 2° to 8° C (36° to 46° F). Solution diluted in D5W is stable for 3 hours at RT (15° to 30° C [59° to 86° F]) and room light or for 24 hours refrigerated at 2° to 8° C (36° to 46° F). Administration must be completed within these times (e.g., 3, 6, or 24 hours based on type of storage and solution used). Bendeka is supplied as a multiple-dose vial that is stable for up to 28 days when stored in its original carton under refrigeration. Manufacturer recommends no more than 6 dose withdrawals from each vial. Treanda Injection must be stored in the refrigerator at 2° to 8° C (36° to 46° F). Solution diluted in NS or D2.5/1/2NS is stable for 2 hours at RT (15° to 30° C [59° to 86° F]) and room light or for 24 hours refrigerated at 2° to 8° C (36° to 46° F). Administration must be completed within these times (e.g., 2 or 24 hours based on type of storage). Treanda for Injection may be stored at CRT. Solution diluted in NS or D2.5/1/2NS is stable for 3 hours at RT (15° to 30° C [59° to 86° F]) and room light or for 24 hours refrigerated at 2° to 8° C (36° to 46° F). Administration must be completed within these times. Discard any unused solution.
Compatibility
Manufacturer states, “Use SWFI for reconstitution (for Treanda for Injection) and then (for both formulations) either NS or D2.5/1/2NS for dilution. No other diluents have been shown to be compatible.”
Treanda injection:
Contains N,N-dimethylacetamide (DMA), which is incompatible with devices that contain polycarbonate or ABS. Devices, including CSTDs, adaptors, and syringes that contain polycarbonate or ABS, have been shown to dissolve when they come into contact with DMA. This incompatibility leads to device failure (e.g., leaking, breaking, or operational failure of CSTD components), possible product contamination, and potential serious adverse health consequences to the practitioner or patient.
Rate of administration
Bendeka:
All indications:
Total daily dose as an infusion equally distributed over 10 minutes.
Treanda:
Chronic lymphocytic leukemia (CLL):
Total daily dose as an infusion equally distributed over 30 minutes.
Non-Hodgkin’s lymphoma (NHL):
Total daily dose as an infusion equally distributed over 60 minutes.
Actions
A bifunctional mechlorethamine derivative. An alkylating agent. Active against both quiescent and dividing cells. Exact mode of action unknown. May lead to cell death by damaging the DNA in cancer cells as well as by disrupting normal cell division. Highly protein bound. Distributes freely in human red blood cells. Extensively metabolized via hydrolytic, oxidative, and conjugative pathways. Excreted in urine and feces.
Indications and uses
Treatment of patients with chronic lymphocytic leukemia (CLL). Study demonstrated a higher rate of overall response and a longer progression-free survival for bendamustine compared with chlorambucil (Leukeran). Effectiveness compared with first-line therapies other than chlorambucil has not been studied. ■ Treatment of patients with indolent B-cell non-Hodgkin’s lymphoma that has progressed during or within 6 months of treatment with rituximab (Rituxan) or a rituximab-containing regimen.
Contraindications
Bendeka:
Known hypersensitivity to bendamustine, polyethylene glycol 400, propylene glycol, or monothioglycerol.
Treanda:
Known hypersensitivity to bendamustine.
Precautions
Follow guidelines for handling cytotoxic agents. See Appendix A, p. 1331. ■ Administered by or under the direction of the physician specialist in a facility equipped to monitor the patient and respond to any medical emergency. ■ Do not use in patients with a CrCl less than 40 mL/min. Use with caution in patients with mild or moderate renal impairment; no formal studies conducted. ■ Use with caution in patients with mild hepatic impairment. Do not use in patients with moderate hepatic impairment (AST or ALT 2.5 to 10 times the ULN and total bilirubin 1.5 to 3 times the ULN) or severe hepatic impairment (total bilirubin greater than 3 times the ULN). No formal studies conducted. ■ Myelosuppression may be severe and require dose delays and/or subsequent dose reductions. Deaths from myelosuppression-related adverse reactions have occurred; see Dose Adjustments. ■ Infections, including hepatitis, pneumonia, and sepsis, have been reported. Has been associated with septic shock and death. ■ Patients treated with bendamustine are at risk for reactivation of infections, including (but not limited to) hepatitis B, cytomegalovirus, Mycobacterium tuberculosis, and herpes zoster; see Monitor. ■ Infusion reactions are common. In rare instances anaphylaxis or anaphylactoid reactions have occurred. Usually occur in the second and/or subsequent cycles of therapy. ■ Tumor lysis syndrome (TLS) has been reported and may occur in the first treatment cycle. S/S are rapid reduction in tumor volume, renal insufficiency, hyperkalemia, hypocalcemia, hyperuricemia, or hyperphosphatemia. May lead to acute renal failure and death. ■ Skin reactions, including rash, toxic skin reactions, and bullous exanthema, have been reported; see Drug/Lab Interactions and Antidote. ■ Premalignant and malignant diseases, including myelodysplastic syndrome, myeloproliferative disorders, acute myeloid leukemia, and bronchial carcinoma have been reported. Causal relationship has not been determined.
Monitor:
Patients should undergo appropriate measures (including clinical and laboratory monitoring, prophylaxis, and treatment) for infection and/or infection reactivation before treatment. ■ Obtain baseline CBC with differential and platelet count. Monitor CBC with differential weekly, and monitor platelet count each cycle. Hematologic nadir usually occurs in the third week. If recovery to recommended values does not occur by the first day of the next scheduled cycle, delay dose until recovery occurs; see Dose Adjustments. ■ Obtain baseline CrCl, AST, ALT, and total bilirubin; repeat as indicated. ■ Monitor closely for S/S of infusion or hypersensitivity reactions (e.g., chills, fever, pruritus, rash). Discontinue bendamustine if a severe reaction occurs. Inquire about possible symptoms that suggest a minor reaction after the first infusion. Consider premedication with antihistamines (e.g., diphenhydramine [Benadryl]), antipyretics (e.g., acetaminophen [Tylenol]), and corticosteroids in patients who have experienced a Grade 1 or 2 infusion reaction; see Antidote. ■ Monitor for S/S of TLS. In patients at risk for TLS, prevention and treatment of hyperuricemia may be accomplished with vigorous hydration. Allopurinol has been used during the beginning of bendamustine therapy; see Drug/Lab Interactions. Monitor uric acid levels. Monitor electrolytes, particularly potassium, and treat as indicated. ■ Monitor patients with skin reactions closely. Withhold or discontinue bendamustine if skin reactions are severe or progressive. ■ Use prophylactic antiemetics to reduce nausea and vomiting and increase patient comfort. ■ Observe for S/S of infection (e.g., fever) or reactivation of infection. Prophylactic antibiotics may be indicated pending results of C/S in a febrile neutropenic patient. ■ Monitor for thrombocytopenia (platelet count less than 50,000 cells/mm3). Initiate precautions to prevent excessive bleeding (e.g., inspect IV sites, skin, and mucous membranes; use extreme care during invasive procedures; test urine, emesis, stool, and secretions for occult blood). ■ Monitor IV site for signs of extravasation during and after administration (e.g., infection, pain, redness, swelling, necrosis); extravasation has resulted in hospitalization.
Patient education:
Avoid pregnancy; nonhormonal birth control is recommended for both men and women throughout treatment and for 3 months after treatment is complete; report a suspected pregnancy immediately. May pose a risk to reproductive capacity in both males and females. ■ Promptly report signs of infection (e.g., chills, fever) or allergic reaction (e.g., dyspnea, itching, rash) and severe or worsening skin reactions, including itching or rash. ■ Frequent laboratory monitoring required. ■ Promptly report IV site burning or stinging. ■ Report other side effects such as nausea, vomiting, or diarrhea. Symptomatic treatment can be provided. ■ May cause fatigue. Avoid driving or operating any dangerous tools or machinery if this side effect is experienced. ■ See Appendix D, p. 1333.
Maternal/child:
Category D: avoid pregnancy; can cause fetal harm. May also cause impaired spermatogenesis, azoospermia, and total germinal aplasia in males. Males and females of childbearing age must use birth control. ■ If the drug is used during pregnancy or if the patient becomes pregnant during therapy, inform the patient of the potential hazard to the fetus. ■ Has the potential for serious side effects; discontinue breast-feeding. ■ Effectiveness for use in pediatric patients not established. Evaluation of one small Phase 1/2 trial suggests that the safety profile in pediatric patients is similar to that seen in adults; see prescribing information.
Elderly:
Side effect profile similar for all age-groups studied.
Chronic lymphocytic leukemia (CLL):
Response to bendamustine was improved in all age-groups over the response to chlorambucil; however, response in the elderly was less than in younger adults. The progression-free survival time was somewhat longer for younger adults compared with those 65 years of age or older.
Non-Hodgkin’s lymphoma (NHL):
Effectiveness and duration of response similar for all age-groups.
Drug/lab interactions
No formal drug interaction studies have been conducted. ■ Active metabolites of bendamustine are formed via cytochrome P450 CYP1A2. Inhibitors of CYP1A2 (e.g., ciprofloxacin [Cipro], fluvoxamine [Luvox]) may increase plasma concentrations of bendamustine and decrease plasma concentrations of active metabolites. Inducers of CYP1A2 (e.g., omeprazole [Prilosec], smoking) may decrease plasma concentrations of bendamustine and increase plasma concentrations of its active metabolites. Use caution or consider alternative treatments if concomitant treatment with CYP1A2 inhibitors or inducers is indicated. ■ Not likely to inhibit metabolism via other selected CYP isoenzymes or to induce the metabolism of substrates of cytochrome P450 enzymes. ■ Cases of Stevens-Johnson syndrome (SJS) and toxic epidermal necrolysis (TEN) have been reported with concomitant administration of allopurinol and other medications known to cause these syndromes (e.g., rituximab [Rituxan]). ■ In vitro data suggest that P-glycoprotein breast cancer resistance protein (BCRP) and/or other efflux transporters may have a role in bendamustine transport.
Side effects
Anorexia, constipation, cough, diarrhea, dyspnea, fatigue, fever, headache, myelosuppression (anemia, febrile neutropenia, leukopenia, lymphopenia, neutropenia, thrombocytopenia), nausea, rash, stomatitis, vomiting, and weight loss were most common. Other side effects reported include asthenia; chills; decreased CrCl; dry mouth; elevated AST, ALT, and bilirubin levels; herpes simplex; hypersensitivity and/or infusion reactions (e.g., anaphylaxis [rare], pruritus, rash); hypertension; hyperuricemia; infections; malaise; malignancies; mucosal inflammation; nasopharyngitis; pneumonia; sepsis; somnolence; stomatitis; TLS; and weakness. Hypersensitivity reactions and fever required study withdrawal in some patients.
Post-marketing:
Anaphylaxis, cardiac disorders (atrial fibrillation, CHF [some fatal], MI [some fatal], palpitation), extravasation resulting in hospitalization, infusion site reactions (irritation, pain, phlebitis, pruritus, swelling), pancytopenia, Pneumocystis jiroveci pneumonia, pneumonitis, Stevens-Johnson syndrome, and toxic epidermal necrolysis.
Overdose:
Ataxia, cardiac arrhythmias, convulsions, respiratory distress, sedation, tremor.
Antidote
Keep physician informed of all side effects and hematologic parameters. Side effects may decrease in severity with reduced dose. Bone marrow depression may require withholding bendamustine until recovery occurs. Administration of whole blood products (e.g., packed RBCs, platelets, leukocytes) may be required. Selected blood modifiers (e.g., erythropoiesis-stimulating agents [ESAs (Aranesp, Epogen, Mircera)], filgrastim [Neupogen, Zarxio], pegfilgrastim [Neulasta], sargramostim [Leukine]) may be indicated to treat bone marrow toxicity. Discontinue the infusion immediately for any life-threatening side effect (e.g., clinically significant bronchospasm, cardiac arrhythmias, severe hypotension). Consider premedication with antihistamines (e.g., diphenhydramine [Benadryl]), antipyretics (e.g., acetaminophen [Tylenol]), and corticosteroids in subsequent cycles in patients who have experienced a Grade 1 or 2 hypersensitivity and/or infusion reaction. Grade 3 or 4 reactions have not typically been rechallenged; consider discontinuing bendamustine. Withhold or discontinue bendamustine if skin reactions are severe or progressive. There is no specific antidote. Supportive therapy as indicated will help sustain the patient in toxicity. ECG monitoring may be indicated to evaluate cardiac side effects.
Bevacizumab 
(beh-vah-SIZZ-ih-mab)
Avastin
Recombinant monoclonal antibody
Angiogenesis inhibitor
Antineoplastic
pH 6.2
Usual dose
Do not begin therapy until at least 28 days after major surgery. Surgical incisions should be fully healed. May be used in combination with other antineoplastic agents or as a single agent. See Dose Adjustments, Monitor, and Precautions. Continue treatment until disease progression or unacceptable toxicity occurs.
Metastatic carcinoma of the colon or rectum:
Administered as an infusion. Recommended doses are either 5 mg/kg or 10 mg/kg every 2 weeks when used in combination with intravenous 5-FU–based chemotherapy. Administer 5 mg/kg when used in combination with bolus IFL (irinotecan [Camptosar], fluorouracil [5-FU], leucovorin calcium). Administer 10 mg/kg when used in combination with FOLFOX4 (fluorouracil [5-FU], leucovorin calcium, and oxaliplatin [Eloxatin]).
Administer 5 mg/kg every 2 weeks or 7.5 mg/kg every 3 weeks when used in combination with a fluoropyrimidine-irinotecan–based or a fluoropyrimidine-oxaliplatin–based chemotherapy regimen in patients who have progressed on a first-line bevacizumab-containing regimen.
Nonsquamous, non–small-cell lung cancer:
15 mg/kg as an infusion every 3 weeks in combination with carboplatin (Paraplatin) and paclitaxel (Taxol).
Glioblastoma:
10 mg/kg every 2 weeks.
Metastatic renal cell carcinoma:
10 mg/kg every 2 weeks in combination with interferon alfa.
Cervical cancer:
15 mg/kg as an infusion every 3 weeks administered in combination with one of the following chemotherapy regimens: paclitaxel (Taxol) and cisplatin, or paclitaxel and topotecan (Hycamtin).
Platinum-resistant recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer:
10 mg/kg every 2 weeks in combination with one of the following chemotherapy regimens: paclitaxel, pegylated liposomal doxorubicin, or topotecan (weekly); or 15 mg/kg every 3 weeks in combination with topotecan (every 3 weeks).
Dose adjustments
No dose adjustments are recommended based on age, gender, or race. ■ No dose adjustments are recommended for patients with impaired hepatic or renal function; bevacizumab was not studied in these patients. ■ Permanently discontinue if GI perforation, fistula formation in the GI tract (e.g., enterocutaneous, esophageal, duodenal, rectal), intra-abdominal abscess, fistula formation involving an internal organ, wound dehiscence (parting of the sutured lips of a surgical wound) or wound healing complications requiring medical intervention, serious bleeding, severe arterial thromboembolic event, life-threatening (Grade 4) venous thromboembolic events (including pulmonary embolism), nephrotic syndrome, hypertensive crisis or hypertensive encephalopathy, or posterior reversible encephalopathy syndrome (PRES) develops; see Monitor and Precautions. ■ Temporarily discontinue if moderate to severe proteinuria (equal to or greater than 2 Gm/24 hr), severe hypertension not controlled with medical management, and/or a severe infusion reaction occurs; see Monitor and Precautions. ■ Withhold bevacizumab for at least 4 weeks before elective surgery (half-life is approximately 20 days but has a wide range). Incision must be fully healed before therapy is resumed.
Dilution
Available in single-use vials containing 100 mg in 4 mL or 400 mg in 16 mL (25 mg/mL). Calculate desired dose and choose the appropriate vial or combination of vials. Withdraw the required volume of bevacizumab and dilute in a total volume of 100 mL of NS.
Filters:
Not required by manufacturer; however, studies using a 0.2-micron in-line filter were done, and drug potency appeared to be maintained.
Storage:
Store in original carton in refrigerator at 2° to 8° C (36° to 46° F). Protect from light. Do not shake or freeze. Diluted solutions may be refrigerated for up to 8 hours. Contains no preservatives; unused portions must be discarded.
Compatibility
Manufacturer states, “Should not be administered with or mixed with dextrose solutions.” Incompatibilities with polyvinylchloride or polyolefin bags have not been observed.
Rate of administration
Do not administer as an IV push or bolus. Must be given as an infusion. Administer following concurrent chemotherapy. Infusion reactions are not common but may occur; see Monitor.
Initial infusion:
A single dose equally distributed over 90 minutes.
Second infusion:
If the initial infusion is well tolerated, the second infusion may be administered equally distributed over 60 minutes.
Subsequent infusions:
If the 60-minute infusion is well tolerated, subsequent infusions may be administered equally distributed over 30 minutes.
Actions
A humanized IgG1 monoclonal antibody produced by recombinant DNA technology. Has antiangiogenesis properties; it binds to and inhibits the biologic activity of human vascular endothelial growth factor (VEGF). The interaction of VEGF with its receptors leads to endothelial cell proliferation and new blood vessel formation. By binding VEGF, bevacizumab prevents the interaction of VEGF with its receptors on the surface of endothelial cells, thus inhibiting the development of new blood vessels around tumors (a tumor-starving mechanism) and resulting in a reduction of microvascular growth and an inhibition of metastatic disease progression. Predicted time to steady-state was 100 days. Half-life is approximately 20 days (range is 11 to 50 days). IgG antibodies may cross the placental barrier and be secreted in breast milk.
Indications and uses
First-line or second-line treatment of metastatic carcinoma of the colon or rectum. Used in combination with intravenous 5-fluorouracil–based chemotherapy (e.g., IFL, FOLFOX4). ■ Second-line treatment of metastatic colorectal cancer in patients who have progressed on a first-line bevacizumab-containing regimen. Used in combination with fluoropyrimidine-irinotecan–based or fluoropyrimidine-oxaliplatin–based chemotherapy. ■ First-line treatment of patients with unresectable, locally advanced, recurrent, or metastatic nonsquamous, non–small-cell lung cancer. Given in combination with carboplatin and paclitaxel. ■ Treatment of glioblastoma in adult patients as a single agent for patients with progressive disease following prior therapy. (Effectiveness is based on an improvement in objective response rate. There are no data showing an improvement in disease-related symptoms or increased survival with bevacizumab.) ■ Treatment of metastatic renal cell cancer. Given in combination with interferon alfa. ■ Treatment of persistent, recurrent, or metastatic carcinoma of the cervix. Given in combination with paclitaxel and cisplatin or paclitaxel and topotecan. ■ Treatment of patients with platinum-resistant recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who received no more than 2 prior chemotherapy regimens. Given in combination with paclitaxel, pegylated liposomal doxorubicin, or topotecan.
Limitation of use:
Bevacizumab is not indicated for adjuvant treatment of colon cancer.
Contraindications
Manufacturer states, “No known contraindications.” However, bevacizumab must be discontinued if GI perforation, wound dehiscence requiring medical intervention, serious bleeding, nephrotic syndrome, or hypertensive crisis develops; see Antidote. ■ Use with caution in patients with known hypersensitivity to murine proteins, bevacizumab, or any of its components. ■ Not recommended for use in patients with recent hemoptysis (greater than or equal to 1/2 teaspoon of red blood). ■ Avoid use in patients with ovarian cancer who have evidence of rectosigmoid involvement by pelvic examination or bowel involvement on CT scan or clinical symptoms of bowel obstruction.
Precautions
Do not administer as an IV push or bolus. Must be given as an infusion. ■ Should be administered by or under the direction of a physician specialist in a facility equipped to monitor the patient and respond to any medical emergency. ■ Has been shown to impair wound healing; withhold bevacizumab for a minimum of 28 days after major surgery; surgical incision must be fully healed. Withhold at least 28 days before elective surgery (half-life is approximately 20 days but has a wide range). Appropriate intervals between surgery and the beginning of bevacizumab therapy and/or the end of bevacizumab therapy and subsequent elective surgery have not been determined. ■ GI perforation with or without fistula formation and/or intra-abdominal abscesses has occurred; deaths have been reported. The majority of cases occurred within the first 50 days of initiation of bevacizumab. Consider GI perforation in any patient with complaints of abdominal pain associated with constipation, fever, nausea, and vomiting. ■ Nongastrointestinal fistula formation has been reported, in some cases with a fatal outcome. Fistula formations involving tracheoesophageal, bronchopleural, biliary, vaginal, renal, and bladder areas have been reported. Most events occurred within the first 6 months of bevacizumab therapy. ■ Necrotizing fasciitis (including fatal cases) has been reported, usually secondary to wound healing complications, gastrointestinal perforation, or fistula formation. ■ Severe or fatal hemorrhage, including CNS hemorrhage, epistaxis, GI bleed, hematemesis, hemoptysis, and vaginal bleeding occurred up to five times more frequently in patients receiving bevacizumab. Do not administer bevacizumab to patients with serious bleeding or hemoptysis. ■ May cause severe hypertension that may be persistent. Treatment is required; see Monitor and Antidote. ■ Posterior reversible encephalopathy syndrome (PRES) has been reported. Onset of symptoms occurred from 16 hours to 1 year after initiation of therapy. May present with blindness and other visual and neurologic disturbances, confusion, headache, lethargy, and seizures. Mild to severe hypertension may be present. MRI is required to confirm diagnosis. Symptoms usually resolve gradually with discontinuation of bevacizumab and treatment of hypertension. ■ Severe neutropenia, febrile neutropenia, and infection with neutropenia have been reported in patients treated with myelosuppressive chemotherapy plus bevacizumab. ■ Proteinuria occurred during studies and progressed to nephrotic syndrome in some patients. Findings consistent with thrombotic microangiopathy have been found on kidney biopsy in some patients. In other patients, proteinuria decreased within several months after therapy was discontinued. Increased serum creatinine levels have occurred and may not return to baseline. ■ CHF has been reported. Incidence is higher in patients receiving chemotherapy plus bevacizumab compared with patients receiving chemotherapy alone. ■ Infusion reactions are infrequent but have occurred. S/S may include chest pain, diaphoresis, Grade 3 hypersensitivity, headache, hypertension, hypertensive crisis associated with neurologic S/S, oxygen desaturation, rigors, and wheezing. ■ Serious and sometimes fatal arterial thromboembolic events (e.g., CVA [stroke], MI, TIA, angina) have been reported. Incidence is greater with bevacizumab given in combination with chemotherapy as compared to those receiving chemotherapy alone. Risk increased in patients with a history of arterial thromboembolism or diabetes and in patients greater than 65 years of age. ■ Venous thromboembolic events (e.g., deep vein thrombosis, intra-abdominal thrombosis, and pulmonary embolism) have been reported. Patients treated for persistent, recurrent, or metastatic cervical cancer with bevacizumab may be at increased risk for venous thromboembolic events; see Antidote. ■ Avastin may cause fetal harm; see Maternal/Child. ■ Bevacizumab increases the risk of ovarian failure (defined as amenorrhea lasting 3 or more months, FSH level equal to or greater than 30 mIU/mL, and a negative serum β-HCG pregnancy test). Recovery of ovarian function occurred in some but not all women following discontinuation of therapy. Long-term effects of bevacizumab exposure on fertility are unknown. ■ A protein substance, it has the potential for producing an immune response. Neutralizing antibodies against bevacizumab have been found using a specific immunosorbent assay; clinical significance is not known. ■ See Drug/Lab Interactions and Antidote.
Monitor:
Obtain baseline BP, CBC with differential and platelets, electrolytes, and urinalysis. ■ Monitor for S/S of an infusion reaction (e.g., chest pain, chills, diaphoresis, headache, hypertension, hypertensive crisis associated with neurologic S/S, oxygen desaturation, wheezing). ■ Monitor V/S and BP at least every 2 to 3 weeks; monitor more frequently in patients with hypertension. ACE inhibitors, beta-blockers, calcium channel blockers, and diuretics may be used to manage hypertension. Continue to monitor BP at regular intervals after therapy is discontinued. ■ Repeat CBC with differential and platelets and electrolytes as indicated. ■ Use of prophylactic antibiotics may be indicated pending C/S in a febrile, neutropenic patient. ■ Monitor for the development or worsening of proteinuria by serial dipstick urinalysis. Patients with a 2+ or greater urine dipstick reading should undergo further assessment with a 24-hour urine collection. Monitor patients with moderate to severe proteinuria until improvement and/or resolution is observed. Repeat urinalyses and/or 24-hour urine collections as indicated. ■ Monitor for S/S of CHF (e.g., cyanosis, dyspnea on mild exertion, edema, fatigue on exertion, hypoxemia, intolerance to cold, jugular venous distension, orthopnea, pulmonary rales, tachycardia, third heart sound). ■ Check surgical wounds for wound dehiscence. ■ Monitor for S/S of any type of bleeding. ■ Monitor for GI perforation or fistula formulation (e.g., abdominal pain, constipation, fever, hypotension, nausea and vomiting). ■ Monitor for S/S of thromboembolic events. ■ See Dose Adjustments, Rate of Administration, Precautions, and Antidote.
Patient education:
May cause fetal harm; avoid pregnancy. Use effective contraception during treatment with and for 6 months after the last dose of bevacizumab; see Maternal/Child. Women should report a suspected pregnancy immediately. ■ Increases risk of ovarian failure and may impair fertility. ■ Full disclosure of health history is imperative. ■ Promptly report any unusual or unexpected symptoms or side effects (e.g., abdominal pain, bleeding from any source, constipation, dyspnea, fever, persistent cough, rigors, sudden onset of worsening neurologic function, vomiting, wound separation). ■ Routine monitoring of BP required. ■ See Appendix D, p. 1333.
Maternal/child:
May cause fetal harm based on findings from animal studies and the drug’s mechanism of action. Females of reproductive potential are advised to use effective contraception during treatment with bevacizumab and for 6 months after the last dose of bevacizumab. Multiple congenital malformations have been observed in rabbits. Animal models link angiogenesis and VEFG and VEFG Receptor 2 to critical aspects of female reproduction, embryofetal development, and postnatal development. ■ May cause ovarian failure in premenopausal women; long-term effects on fertility are unknown. ■ Discontinue breast-feeding during treatment with bevacizumab and for a prolonged period following treatment (half-life 20 days [range 11 to 50 days]). ■ Safety and effectiveness for use in pediatric patients (including glioblastoma) not established. Nonmandibular osteonecrosis has been reported in pediatric patients less than 18 years of age who have received bevacizumab. Bevacizumab is not approved for this patient population. ■ Dose-related physeal dysplasia (variations in the growth plate) occurred in tested juvenile monkeys; partially reversible after therapy is discontinued.
Elderly:
Overall survival was similar compared to younger adults; however, the incidence of some side effects was increased (e.g., anemia, anorexia, arterial thromboembolic events [e.g., CVA (stroke), MI, TIA, angina], asthenia, CHF, constipation, deep thrombophlebitis, dehydration, diarrhea, dyspepsia, edema, epistaxis, fatigue, GI hemorrhage, hypertension, hypokalemia, hyponatremia, hypotension, ileus, increased cough, leukopenia, nausea and vomiting, proteinuria, sepsis, venous thromboembolic events [e.g., deep vein thrombosis, intra-abdominal thrombosis, pulmonary embolism], voice alteration).
Drug/lab interactions
Drug interaction studies have not been completed. ■ Has been administered concurrently with a regimen of irinotecan, 5-fluorouracil, and leucovorin. Studies indicate no significant effect of bevacizumab on the pharmacokinetics of irinotecan or its active metabolite SN-38. ■ Has been administered with carboplatin, paclitaxel, and interferon alfa. ■ Several cases of microangiopathic hemolytic anemia (MAHA) have been reported in patients with solid tumors who are receiving concomitant therapy with bevacizumab and sunitinib malate (Sutent). This combination therapy is not approved and not recommended. ■ ACE inhibitors, beta-blockers, calcium channel blockers, and diuretics have been coadministered to control hypertension.
Side effects
The most common side effects include back pain, dry skin, epistaxis, exfoliative dermatitis, headache, hypertension, lacrimation (excess), proteinuria, rectal hemorrhage, rhinitis, and taste alteration. Major, dose-limiting, and potentially life-threatening side effects include arterial thromboembolic events (e.g., angina, cerebral infarction, MI, TIA), bleeding episodes (e.g., CNS hemorrhage, epistaxis [severe], GI hemorrhage, hemoptysis, vaginal bleeding), GI perforations, hypertensive crises, infusion reactions, nongastric intestinal fistula formation, posterior reversible encephalopathy syndrome (PRES), proteinuria and/or nephrotic syndrome, surgery and wound healing complications, and venous thromboembolic events. Other reported side effects included abdominal pain, abnormal gait, alopecia, anorexia, anxiety, asthenia, bilirubinemia, CHF, colitis, confusion, constipation, cough, dehydration, diarrhea, dizziness, dry mouth, dysarthria, dyspepsia, dysphonia, dyspnea, edema, fatigue, flatulence, hematologic toxicity (e.g., anemia, leukopenia, neutropenia, thrombocytopenia), hyperglycemia, hypertension, hypoalbuminemia, hypokalemia, hypomagnesemia, hyponatremia, hypotension, ileus, increased serum creatinine, infection, myalgia, nail disorder, nausea, pain, palmar-plantar erythrodysesthesia syndrome, pneumonitis, renal thrombotic microangiopathy (manifested as severe proteinuria), sensory neuropathy, sepsis, skin discoloration, skin ulcer, stomatitis, syncope, urinary frequency and urgency, venous thromboembolic events (e.g., deep vein thrombosis, intra-abdominal thrombosis, pulmonary embolism), voice alteration, vomiting, weight loss.
Post-marketing:
Anastomotic ulceration, acute hypertensive episodes, gallbladder perforation, GI fistula formation (e.g., gastrointestinal, enterocutaneous, esophageal, duodenal, rectal), GI perforation, GI ulcer, hepatobiliary disorders, intestinal necrosis, intra-abdominal abscess, mesenteric venous occlusion, nasal septum perforation, necrotizing fasciitis, nonmandibular osteonecrosis, osteonecrosis of the jaw, ovarian failure, pancytopenia, polyserositis, pulmonary hypertension.
Antidote
Keep physician informed of all side effects. May constitute a medical emergency or will be treated symptomatically as indicated. Permanently discontinue if any of the following develop: GI perforation, fistula formation in the GI tract (e.g., enterocutaneous, esophageal, duodenal, rectal), intra-abdominal abscess, fistula formation involving an internal organ, formation of a tracheoesophageal fistula or any Grade 4 fistula, wound dehiscence or wound healing complications requiring medical intervention, necrotizing fasciitis, serious bleeding requiring medical intervention, nephrotic syndrome, a severe arterial thromboembolic event, life-threatening (Grade 4) venous thromboembolic events (including pulmonary embolism), PRES, hypertensive crisis, or hypertensive encephalopathy. Treat these side effects aggressively; see Precautions and Monitor. Discontinue bevacizumab for severe infusion reactions and treat as indicated (e.g., epinephrine, diphenhydramine [Benadryl], IV fluids, oxygen). Data on rechallenge not available. Temporarily discontinue if moderate to severe proteinuria (equal to or greater than 2 Gm/24 hr) occurs. Resume therapy when proteinuria is less than 2 Gm/24hr; see Monitor and Precautions. Temporarily discontinue if severe hypertension not controlled with medical management occurs; see Monitor and Precautions. Thromboembolic events (e.g., deep vein thrombosis, intra-abdominal thrombosis, pulmonary embolism) were treated with full-dose warfarin (Coumadin) during clinical trials. Monitor INR closely. Bleeding occurred in patients with elevated INRs; relationship to bevacizumab not determined. Withhold bevacizumab for at least 28 days before elective surgery (half-life is approximately 20 days but has a wide range). Incision must be fully healed before therapy is resumed.
Bivalirudin
(by-val-ih-ROO-din)
Angiomax
Anticoagulant
pH 5 to 6
Usual dose
Initiate just before percutaneous coronary intervention (PCI) or percutaneous transluminal coronary angioplasty (PTCA). Given in combination with aspirin.
Aspirin:
300 to 325 mg before PCI and daily thereafter.
For patients who DO NOT HAVE heparin-induced thrombocytopenia (HIT) or heparin-induced thrombocytopenia and thrombosis syndrome (HITTS):
Bivalirudin:
Begin with an IV bolus dose of 0.75 mg/kg. Follow immediately with an infusion at 1.75 mg/kg/hr for the duration of the PCI/PTCA procedure. Perform an ACT 5 minutes after the bolus dose has been administered. An additional bolus dose of 0.3 mg/kg should be given if needed (e.g., ACT less than 225 seconds). Administration with a glycoprotein IIb/IIIa inhibitor (e.g., abciximab [ReoPro], eptifibatide [Integrilin], tirofiban [Aggrastat]) should be considered in any of the following circumstances:
• Decreased TIMI (0 to 2) or slow reflow (Thrombolysis in Myocardial Infarction [TIMI])
• Dissection with decreased flow
• Persistent residual stenosis
• Abrupt closure, clinical instability
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


