Chapter 6 Acute HIV infection
Pathophysiology
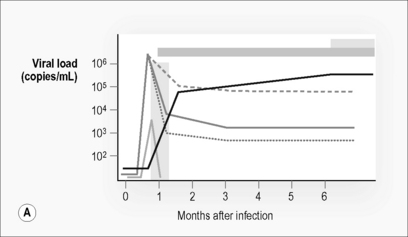
By the time a patient presents with clinical manifestations of acute HIV infection, even in those with negative or indeterminate serology, a whole series of virological and immunological events have already occurred (Fig. 6.1). The so-called window period, despite being apparently silent on diagnostic tests, is neither immunologically nor virologically silent. Insight into many of these earliest events has been gained from animal models such as SIV infection of rhesus macaques as these events are impossible to study in humans for a range of practical and ethical reasons.
Most infections occur at mucosal membranes. After crossing epithelial barriers, the initial cells infected by virus are both dendritic cells and CD4 T cells that populate the mucosal tissues of the genitourinary and/or colonic mucosa. Dendritic cells are capable of harboring and transporting the virus to lymphoid tissue with or without being productively infected [1]. However, whether infected or just transporting the virus, these cells are capable of transferring virus to multiple CD4 T cells as they fulfill their normal role of antigen presentation to T cells. This interaction simultaneously triggers an immune response consisting of both T cell (involving activation of both CD4 and CD8 T cells) and antibody responses while facilitating infection of responding CD4 T cells. This process drives preferential infection and subsequent death of HIV-specific CD4 T cells, resulting in the early deletion of these critical cells from the host’s immune response to the virus [2]. After infection of lymphoid tissue there is subsequent, rapid dissemination of the virus to multiple tissues including the central nervous system.
CD4 T cell responses
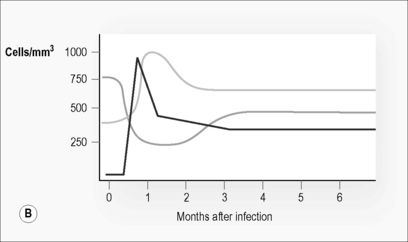
As with any primary immune response, the adaptive immune response takes time to develop. The virus replicates relatively unchecked during this period. Plasma viremia is detectable and viral load increases exponentially (Fig. 6.1). At primary infection in SIV-macaque models there is massive infection and depletion of CD4 T cells, particularly those at tissue sites such as lymphoid tissue, gut, and genitourinary tracts [3, 4]. Similar processes appear to be occurring in humans early in HIV infection [5, 6]. This depletion in tissues is reflected in a reduction in CD4 T cell count in peripheral blood.
In humans, HIV-specific CD4 T cell responses are detectable early in infection [7, 8]. Initially, these consist of cells capable of producing both IL-2 and IFN-γ and of proliferating in response to HIV antigens, but these cells rapidly lose their ability to proliferate and produce IL-2 in the majority of patients (Fig. 6.1 C) [7, 9, 10]. The exception appears to be in those individuals that control virus replication. In the remainder of patients HIV-specific CD4 T cells are detectable, but consist almost entirely of cells capable of producing IFN-γ without proliferative or IL-2-producing capacity (Fig. 6.1 C). This depletion occurs in both peripheral blood and tissues.
T cells homing to peripheral lymphoid tissue such as the gut and genitourinary tracts preferentially express the chemokine receptor CCR5. The preferential depletion of these cells may be due to selective infection and cytolysis due to a heightened susceptibility to infection by the virus strain associated with transmission [11, 12]. Preferential infection of CD4 T cells specifically responding to HIV infection results in loss of these cells and almost certainly accounts for the pathognomic phenomenon of anergy to HIV antigens seen later in HIV infection [2]. Therapy initiated early can preserve this responsiveness and may limit the depletion from and support the repopulation of these cells at mucosal sites [5, 6].
CD8 T cell responses
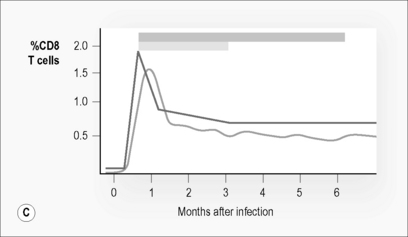
In contrast to the decrease in CD4 T cell numbers, CD8 T cell numbers rise dramatically during acute infection (Fig. 6.1B). This increase consists almost entirely of activated T cells [13]. Many of these activated, proliferating, and expanding cells are cytotoxic T cells (CTL) specifically targeted to HIV (Fig. 6.1 C) [14, 15]. Their ability to lyse infected targets and prevent infection of new cells is thought to limit viral expansion, halting the exponential increase in plasma viral load that follows initial infection and to at least partially determine the eventual viral set point. While CD8 CTL are detectable, a second set of CD8 T cells capable of suppressing viral replication through secretion of a range of soluble factors, including MIP1-α, MIP1-β, RANTES, and an as yet unidentified cytokine called CAF, is also detectable during acute HIV-infection [16]. Deliberate depletion of CD8 T cells from SIV-infected macaques at primary infection results in increases of viral load set point and accounts for more rapid disease progression [17].
In peripheral blood, HIV-specific CD8 T cells are among the first immune responses detectable, usually during the clinical presentation of acute infection. In macaques, SIV-specific CD8 T cells can be detected within 8 days of infection [18]. The initial response is usually highly focused on a limited number of high-avidity epitopes. The response broadens with time. Responses to a range of HIV proteins, including regulatory and accessory proteins such as Tat and Nef, are present [19, 20].
CD8 CTL responses place pressure on the virus. Adaptive evolution of the virus to these responses occurs early, allowing selection of immune escape variants capable of evading CTL responses within several weeks of infection [21, 22].
Antibody responses
Although antibodies to various HIV proteins, particularly p24 and envelope proteins, are detectable soon after infection, most of these antibodies play no clear role in control of infection. The majority of the antibody response is to internal proteins. Of the antibodies directed at the envelope proteins, the overwhelming majority are not neutralizing in nature [23]. Neutralizing antibodies, capable of preventing viral entry and therefore new infection, are slow to arise and are usually detectable not less than 4–8 weeks after infection [24]. When neutralizing antibodies do occur they place pressure on the virus. The virus responds to this pressure, rapidly adapting through the generation of escape variants [25, 26]. New rounds of neutralizing antibodies are also delayed in their development.
Innate immune responses
The role of innate immune responses in primary infection is still not clearly understood. Dendritic cells play a role in the transport of virus to lymphoid tissue, and can act as a “Trojan horse,” transmitting captured virus to CD4 T cells, assisting in the dissemination of infection. However, dendritic cells play a critical role in the control of infection as they are essential for cross-priming and induction of CD8 CTL responses as well as CD4 T cell responses [1]. While myeloid dendritic cell numbers are maintained, plasmacytoid dendritic cell numbers are depleted during primary infection [27].
Natural killer (NK) cells are a significant component of the innate immune response in the early control of viral infections. NK cells increase in number during acute HIV infection prior to seroconversion and before significant CD8 T cell responses are evident [28]. However, as viral replication increases, NK cell responses become impaired [29].
Immune and virological outcomes at resolution of primary infection
Upon onset of these HIV-specific immune responses viral load declines. CD4 T cell counts partially recover but do not return to normal levels. CD8 T cell numbers drop but remain elevated well above the normal range (Fig. 6.1). These changes maintain a reversed CD4:CD8 T cell ratio, despite partial normalization of CD4 T cell counts. Markers of immune activation particularly those expressed on the surface of CD8 T cells (e.g. CD38), remain substantially increased. Plasma viral load reaches a steady state or set point approximately 3–6 months post-infection [30]. This level reflects a balance between the pathogenicity and replication fitness of the virus and the effectiveness of the host’s initial immune response. This level predicts long-term disease outcome (Fig. 6.1) [31].
Virus
Furthermore, the virus appears highly homogenous, shortly after transmission. Two factors may contribute to homogeneity of transmitted strains. The first is the concept of a molecular sieve, where the process of transmission selects for the variants most capable of transmission from among the swarm of quasispecies in the transmitting host. Certain envelope variants appear to have an advantage during the transmission process. The selection of CCR5 tropic variants is the clearest example. More recently, in subtype C virus, variants carrying env sequences with shorter V1–V4 intervals and reduced numbers of glycosylation sites appear to be preferentially transmitted [32]. This molecular sieve is most obvious in heterosexual transmission where the majority of transmissions are by a single viral quasispecies that is most often not the dominant quasispecies in the transmitting host. The restrictions on transmission are less stringent when they occur in men who have sex with men, in the presence of intercurrent sexually transmitted infections, and in intravenous drug users where a larger proportion of transmissions involve multiple quasispecies [33–35]. These observations suggest that intact mucosal surfaces impose a strong selection pressure on the virus.
Additionally, homogeneity of transmitted strains may be contributed to by rapid viral adaptation to its new host post-transmission. Post-transmission, the virus tends to revert from mutations that were advantageous in the original host towards wild type. This process of reversion affects both drug resistance and immune escape mutations [36, 37]. The rate of reversion inversely correlates with the fitness cost to the virus in its new host with reversion of non-advantageous mutations in the new host occurring very quickly.
Despite these processes, a range of adaptive mutations within the transmitted virus may be maintained. Horizontal and vertical transmission of both drug resistance mutations and immune escape mutations are well documented [38, 39]. However, thereafter, mutations reflecting adaptation to the new host also occur rapidly with mutations allowing escape from both CTL and neutralizing antibody pressure arising rapidly [21, 22, 26]. The virus adapts to the altered pressures placed upon it, with selection of the most replication-competent variants occurring rapidly. Variation can occur in all genes. The reasons for differences in the rate of variation relate to varying levels of gene plasticity and differences in the pressures applied.
Co-infection and superinfection
As explained above, simultaneous co-infection with several quasispecies occur in a significant minority of cases especially in the presence of impaired mucosal barriers. In addition superinfection with a second virus within the first 2 years post initial infection has been reported with increasing frequency, at least in non-B subtype infections. Super-infection, in B subtype infections, appears rare or at least uncommon [40–42]; however, in areas where micro-epidemics of different subtypes intersect, superinfection appears more common [43, 44]. The presence of more than one viral quasispecies may be advantageous to the virus, allowing it to evolve more quickly within a host through recombination events.
Clinical Manifestations
The rate of recognition of the clinical manifestations of primary infection varies markedly. An acute illness associated with recent infection with HIV-1 can be identified in the majority of individuals with reported rates ranging from 50 to 90% [45–48]. Initial descriptions described the illness as resembling infectious mononucleosis, with the major manifestations being fever, pharyngitis, and adenopathy [45], but further studies have demonstrated that using this triad alone to describe the clinical manifestations is restrictive [48]. Symptoms associated with initial infection and subsequent seroconversion can vary from completely absent through to an acute debilitating illness requiring hospitalization. The rate of identification is dependent on high levels of clinical suspicion, experience with making the diagnosis, the availability of medical resources, and suspicion on the part of the patient. Although the overwhelming majority of reports of this illness have been in the context of the developed world with subtype B infections, similar manifestations have been reported with other viral subtypes in developing world settings. Similarly, although most reported series also arise from cohorts where the major mode of transmission is sexual, similar prevalence of symptomatic illness and similar clinical manifestations have been reported in intravenous drug users [9, 49]. In adults, clinical manifestations and severity are not dependent on age, sex, race, or geographical factors. Although no concurrent studies have been performed, a study in a US population showed that 49% sought medical attention for symptoms [50], while up to 44% of African women took some time off work due to primary infection symptoms [51]. Although published reports are sparse, similar manifestations are seen in adolescents [52].
The time from exposure to illness is typically 2–4 weeks (range: 6–42 days) [45–48], but there are rare, isolated reports of delayed seroconversion of up to 12 months post-exposure. The acute illness typically lasts approximately 3 weeks and is of rapid onset. In the main, the symptoms are self-limiting. However, up to 20% of cases can require hospitalization or be associated with the presence of opportunistic infections like candidiasis, herpes zoster, cryptococcosis, and Pneumocystis jiroveci pneumonia. The likelihood of primary infection being complicated by an opportunistic infection is related to the extent of CD4 T cell depression [53].
The main clinical manifestations are fever, pharyngitis, adenopathy, rash, myalgia or arthralgia, headaches, and fatigue or asthenia. The pharyngitis is non-exudative and the tonsils are not coated. The rash is classically maculopapular, symmetrical with lesions 0.5–1 cm in diameter affecting face and or trunk, but may also affect the hands including the palms. Other manifestations include mouth ulcers and gastrointestinal upset including diarrhea, odynophagia, anorexia, abdominal pain, and vomiting. Headaches can be associated with retro-orbital pain exacerbated by movement of the eyes and meningitic or encephalitic symptoms and signs. Lymphadenopathy tends to be more common in the cervical region but can affect axillary and inguinal regions [45–48, 53].
The originally described triad of dominant manifestations—fever, pharyngitis, and lymphadenopathy—occurs in a significant minority of patients. Although fever is the most common manifestation, it occurs in less than three-quarters of patients. In the absence of a typical mononucleosis-like presentation, fever is most commonly associated with headache, oral ulceration, and/or abdominal pain. In the absence of fever the most common manifestations are pharyngitis, lethargy, myalgia, rash, headache, and adenopathy [48]. Lymphadenopathy may be slow to resolve, persisting well after the resolution of other manifestations.
The severity of the illness appears to impact on long-term outcome, with greater severity predicting more rapid progression to disease. More rapid CD4 T cell count declines have been documented in those who present to a physician [54]. The presence of candidiasis, neurological involvement, or a prolonged illness lasting more than 14 days is associated with a worse prognosis [48].
Co-infection with other viruses, such as cytomegalovirus (CMV), or other sexually transmitted infections (STIs), occurs. These co-infections can make the clinical presentation more complicated [55]. Co-infection with other viruses such as herpes viruses, hepatitis B or C virus, or other STIs such as chlamydia or syphilis must be considered in the diagnostic work-up.
As stated, race, mode of acquisition, or viral subtype does not appear to impact upon the severity of the illness. Pre-existing impaired immune responses, as demonstrated by low pre-existing CD4 T cell count, low CD4:CD8 T cell ratios, or impaired delayed-type hypersensitivity reactions, are associated with increased risk of a symptomatic illness, as is transmission from an index case with advanced HIV disease [56, 57].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


