- Maintaining normal sodium and water balance
- Maintaining normal potassium balance
- Poor blood sampling/transport can affect plasma potassium
- Causes and effects of raised plasma/serum sodium
- Causes and effects of reduced plasma/serum sodium
- Causes and effects of raised plasma/serum potassium
- Causes and effects of reduced plasma/serum potassium
The most frequently requested blood chemistry test is Urea and Electrolytes (U&E). This is not one but five tests performed simultaneously on the serum or plasma recovered from one sample of blood. These tests are: serum or plasma concentration of sodium, potassium, bicarbonate, urea and creatinine. The first three are electrolytes, that is positively or negatively charged ions in solution. This chapter focuses on the positively charged ions (cations), sodium (Na+) and potassium (K+). Bicarbonate, a negatively charged ion (anion), is considered in Chapter 7. Urea and creatinine are the subject of Chapter 5.
Physiology – sodium and water balance
[Note: An understanding of sodium and water balance requires outline knowledge of kidney function and the process by which urine is formed from filtered blood. This topic is not addressed here but in Chapter 5, and readers may wish to refer there for clearer understanding of some aspects of this section.]
Sodium is required for nerve cell conduction and bone formation but the principle physiological significance of sodium is that sodium concentration of extracellular fluid determines its volume, which in turn determines blood volume and thereby blood pressure. This function of sodium reflects the interrelatedeness of sodium and water metabolism. As will become clear monitoring water intake and output (fluid balance) is as important as measuring sodium concentration for elucidating cause and monitoring treatment of abnormal plasma sodium concentration.
Distribution of body water
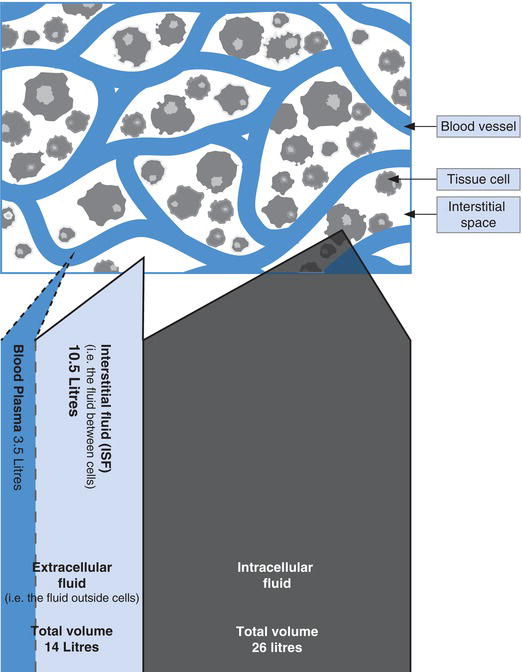
Around 60% of human body weight is water; for an average adult weighing 70 kg this represents close to 40 L of water. Approximately 26 L is contained in the totality of cells within the body, this is called the intracellular fluid (ICF), and 14 L is outside cells and called the extracellular fluid (ECF). The ECF comprises approximately 3.5 L of plasma, the fluid (non-cellular) part of blood contained within the vascular system, and 10.5 L of interstitial (or extravascular) fluid, which fills the microscopical space between tissue cells (Figure 4.1).
It is vital for health that both the total amount of water in the body and its distribution between these compartments is constant. The passage of water across cell membranes, that is between the ECF and ICF, is dependent on the osmolarity on either side of the cell membrane; so long as this is equal, water will not pass and the volumes of each compartment are maintained. In health both plasma (ECF) osmolarity and ICF osmolarity is maintained within the range 280–295 mOsmol/kg.
How does sodium determine ECF volume?
The osmolarity of any solution is determined by the total concentration of dissolved solutes. Because they are present in relatively high concentration in body fluids (i.e. ECF and ICF), compared with other solutes, sodium and potassium are the major determinants of ECF and ICF osmolarity respectively. Figure 4.2 describes the distribution of electrolytes between the ECF and ICF.
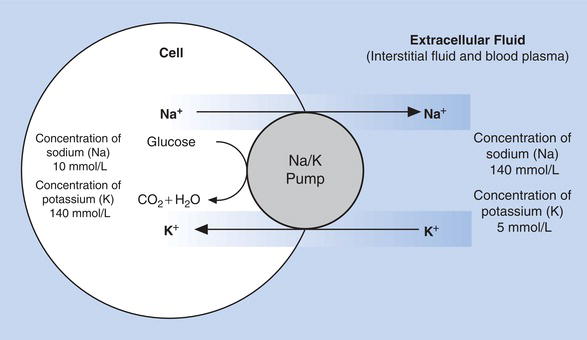
Within cells, the predominant cation is potassium; here there is relatively low concentration of sodium. By contrast, the ECF has a high concentration of sodium and relatively low potassium concentration; these differing electrolyte concentrations on either side of the cell membrane must be maintained by active transport. This active transport is achieved by the so called sodium–potassium pump (Figure 4.3), an energy requiring system present in the membrane of all cells that continuously ‘pumps’ sodium out of cells as potassium is ‘pumped’ in.
Without such active transport, sodium and potassium would diffuse passively across cell membranes until concentrations of ECF and ICF were equal. The active transport of sodium out of cells ensures its high concentration in the ECF, and therefore its predominant effect on osmolarity of the ECF. Since osmolarity determines the distribution of water between the ICF and ECF, and sodium concentration is the principal determinant of ECF osmolarity, it follows that plasma (i.e. ECF) sodium concentration is a major determinant of ECF volume and therefore blood volume.
Control of water balance
To avoid dehydration or overhydration, water intake and output must be the same. A minimum urine volume (i.e. water loss) of 500 ml per day is required for excretion of the waste products of metabolism by the kidneys. To this must be added the water lost via the lungs in expired air (around 400 ml), via the skin in sweat (around 500 ml) and in faeces (around 100 ml). Thus a minimum of 1500 ml of water is normally lost from the body each day. Approximatley 400 ml of water is produced by the body each day, a by-product of cellular metabolism. To maintain normal balance then, a minimum intake of around 1100 ml is required. In practice, fluid intake is greater than this minimum, but the kidneys are easily able to excrete an increased volume of water, to balance intake. In fact on average most people excrete 1200–1500 ml of urine every day, and the kidneys have the capacity to produce a urine volume far in excess of this, if required.
Figure 4.2 Comparison of the normal solute concentration in ECF and ICF.
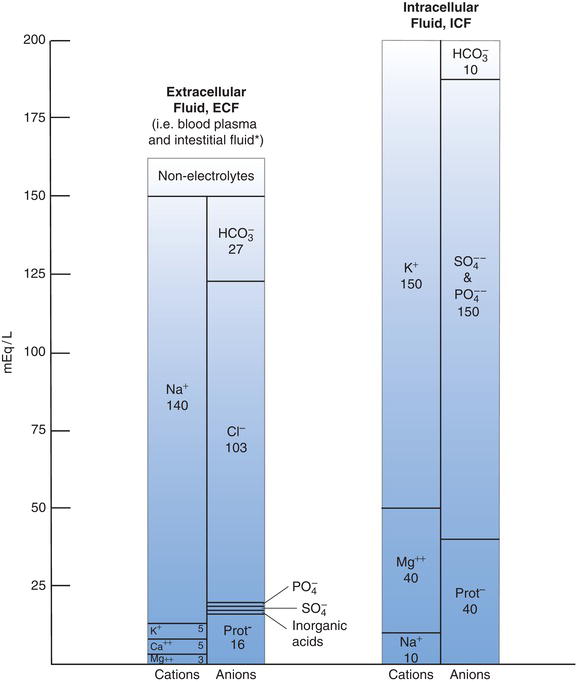
Notes: 1. mEq/L = mmol/L for monovalent ions (e.g. sodium, Na and Potassium, K) but mEq/L must be divided by 2 to convert mmol/L for divalent ions (e.g. Calcium, Ca and Magnesium, Mg).
2. Figures for ECF refer specifically to blood plasma; interstitial fluid very similar except it has lower protein and higher chloride concentration.
Figure 4.3 Maintenance of differential sodium and potassium concentration between cells and surrounding extracellular fluid.
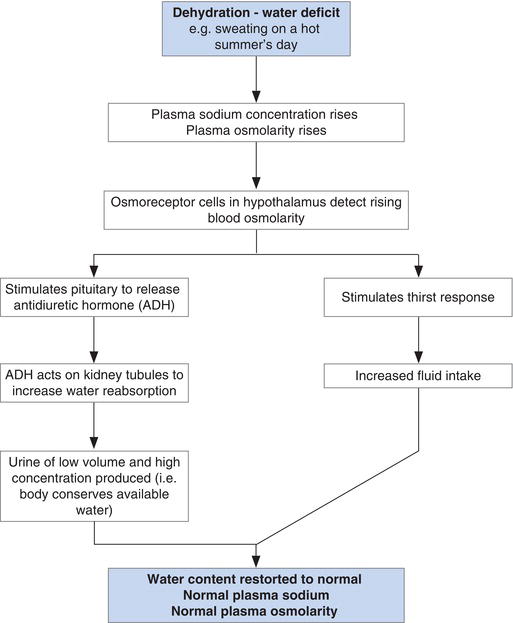
Water intake and urine loss is controlled by plasma osmolarity. If, for example, water is being lost from the body without adequate replacement, the ECF volume decreases and osmolarity rises. This causes water to pass from cells into the ECF, restoring ECF volume and osmolarity to some extent. This internal movement of water can however only be a short-term corrective as cells become relatively dehydrated. More water is required.
The normal response to this water deficit is described in Figure 4.4. As blood with plasma of high osmolarity passes through the hypothalamus in the brain, specialised cells called osmoreceptors sited there respond to increased osmolarity with two simultaneous outcomes: the thirst response is invoked; and the pituitary gland secretes the antidiuretic hormone, arginine vasopressin (AVP). Thirst of course induces increased water intake. AVP conserves body water by its action on the kidney; specifically it increases water reabsorption to blood from the distal tubules and collecting ducts of the kidney. The result of AVP action is excretion of concentrated urine of relatively low volume. The net effect of this dual response is more water in the ECF and restoration of normal plasma osmolarity.
Conversely, if fluid (water) is drunk in excess of current requirement, ECF osmolarity falls and osmoreceptors remain unstimulated. Consequently there is no sensation of thirst and no AVP secretion/action. The net effect is that dilute urine of relatively high volume is excreted, and the effective water overload is corrected; normal plasma osmolarity is restored.
It must be remembered that around 8000 ml of water is secreted into the gastrointestinal tract each day as saliva, gastric juice, bile, pancreatic and intestinal juice. In health 99% of this water is reabsorbed from the gastrointestinal tract and just 100 ml is lost in faeces each day. However, failure to conserve the water contained in these secretions – evident symptomatically as diarrhoea and/or vomiting – can result in severe water imbalance. Maintenance of normal water balance is dependent then on:
- An intact thirst response, which requires consciousness.
- A normally functioning hypothalamus and pituitary gland.
- Normally functioning kidney.
- Normally functioning gastrointestinal tract.
Figure 4.4 Normal water balance is dependent on intact hypothalamic–pituitary axis: adequate ADH, thirst response and renal function.
Control of sodium balance
Just as health demands a balance between water intake and loss, the same is true of sodium. A human adult normally contains around 3000 mmol of sodium, most of which, as we have seen, is present in the ECF (i.e. blood plasma and interstitial fluid) at a concentration of around 140 mmol/L.
A minimum of 10 mmol/day is lost in urine, sweat and faeces and this must be replaced to remain in balance. In fact, we ingest far more than this minimum: the average diet contains on average 100–200 mmol/day, mostly as salt flavouring (a teaspoon of table salt contains 100 mmol sodium). Excess sodium is excreted by the kidneys, in urine. It is kidney regulation of sodium excretion that ensures normal sodium balance, despite wide variation in intake.
Most (90–95%) of the sodium filtered at the glomerulus is reabsorbed to blood during passage of the ultrafiltrate through the proximal convoluted tubule and ascending limb of Henle. By the time the ultrafiltrate arrives in the distal convoluted tubule just 5–10% of sodium filtered at the glomerulus remains. The fate of this sodium, that is whether it is excreted in urine or reabsorbed to blood, is dependent on the blood concentration of the adrenal hormone, aldosterone. This hormone acts on the cells of the distal tubule and collecting duct to enhance sodium reabsorption in exchange for potassium or hydrogen ions. Thus in the presence of high levels of aldosterone most of the remaining sodium in the distal tubule is reabsorbed; if aldosterone concentration is low no more is reabsorbed and urine of relatively high sodium concentration is excreted.
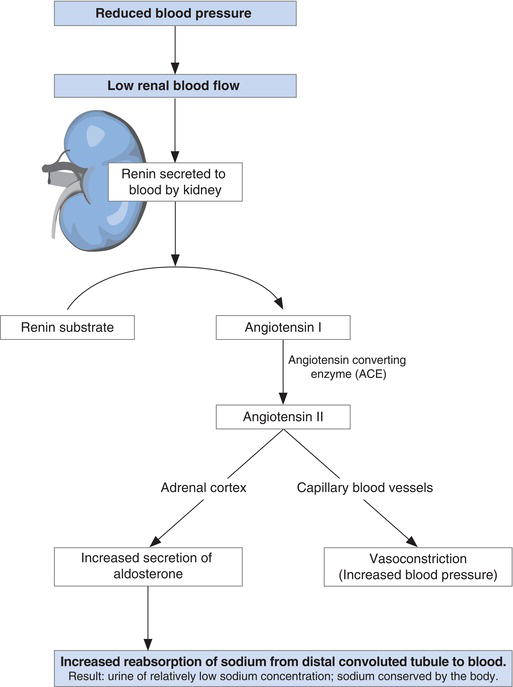
Aldosterone secretion by the adrenal cortex is controlled in turn by the renin–angiotensin system (Figure 4.5). Renin is an enzyme produced by and secreted from the cells of the juxtaglomerulus of the kidney when blood flow through the glomerulus is reduced. Since the rate of blood flow through the kidneys (indeed any organ) is dependent on blood volume and therefore sodium concentration, it follows that renin is secreted from the kidneys when plasma sodium concentration is falling.
Renin enzymically splits angiotensinogen, a protein present in blood; one of the products is a small peptide of ten amino acids called angiotensin I. A second enzyme derived predominantly from the lungs, called angiotensin converting enzyme (ACE) then splits two amino-acids from angiotensin I. The remaining eight amino-acid peptide is the hormone angiotensin II. This hormone has two important effects:
- It causes capillary blood vessels to narrow (a process called vasoconstriction), increasing blood pressure, and thereby increasing renal blood flow.
- It stimulates the cells of the adrenal cortex to synthesise and secrete aldosterone, the effect of which as described here is reabsorption of sodium, and thereby restoration of normal plasma sodium.
Two additional hormones, atrial natriuretic peptide (ANP) and brain natriuretic peptide (BNP) released by the cells of the heart in response to atrial and ventricular stretch, have an antagonistic effect to that of aldosterone. The effect of rising blood levels of ANP and BNP is reduced sodium reabsorption from the distal tubule and excretion of urine with relatively high sodium concentration.
Around 1500 mmol of sodium is excreted into the gastrointestinal tract along with water every day (as already discussed). Normally all – save around 10 mmol which is excreted in faeces – is reabsorbed to blood. Failure of gastrointestinal sodium reabsorption inevitably leads to potential sodium deficit, which becomes real if renal compensation is incomplete.
To summarise then sodium balance is chiefly dependent on:
- Normal kidney function.
- Appropriate secretion of aldosterone by the adrenal cortex gland (which in turn is dependent on an intact renin–angiotensin axis).
- Normal gastrointestinal function.
Normal physiology – potassium
Potassium is required for the action of many metabolic enzymes, electrical transmission of nerve impulses and muscle contraction. Virtually all of the body’s 3000 mmol of potassium is contained within cells. Just 0.4% is in plasma where it can be measured, so that plasma potassium is a poor indicator of total body potassium. The maintenance of normal plasma potassium concentration, which is essential for health, depends on maintaining overall potassium balance.
Normal control of potassium balance
A minimum of around 40 mmol of potassium is lost from the body every day in urine, faeces and sweat. This must be replaced in order to maintain balance. Normal dietary intake is of the order of 100 mmol/day, obtained from citrus fruits and bananas, leafy vegetables, potatoes and bread. The kidneys ensure that sufficient potassium is excreted in urine to match that ingested. As with sodium, most of the potassium filtered at the glomerulus of the kidney is reabsorbed in the first (proximal) part of the kidney tubule. Fine regulation occurs in the distal part of the tubule and collecting ducts. Potassium can be either secreted from blood into the tubule in exchange for sodium ions, or reabsorbed to blood at this point. As has already been made clear sodium–potassium exchange is enhanced by the renin–angiotensin–aldosterone axis, so that under the influence of aldosterone, as sodium is reabsorbed to blood, potassium is lost in urine.
The amount of potassium lost in urine is also affected by the kidneys’ role in maintaining the pH of blood within normal limits. One of the mechanisms for preventing blood becoming too acidic is renal excretion of excess hydrogen ions in urine. This hydrogen ion excretion occurs by exchange with sodium ions in the distal tubule. This means that in acidosis less sodium is available for exchange with potassium and consequently relatively less potassium is excreted in urine. As will become clear, there are other ways in which potassium balance is affected by disturbance in acid-base balance.
Around 60 mmol of potassium is normally secreted into the gastrointestinal tract every day but most is reabsorbed; just 10 mmol/day is lost in faeces. Potassium deficit can occur if this gastrointestinal reabsorption is defective.
Movement of potassium across cell membranes
As already described the high concentration of ICF potassium and low concentration of ECF (plasma) potassium is maintained by the sodium–potassium pump. Increased and decreased activity of this pump can have profound effect on the plasma potassium concentration, as potassium shifts between the ECF and ICF. In addition, since hydrogen ions compete with potassium for exchange across the cell membranes, disturbances of acid base-balance may affect the plasma potassium concentration. An abnormal increase or decrease in plasma potassium concentration does not necessarily mean an overall body deficit or excess, it may simply reflect a shift of potassium into or out of cells.
Maintenance of a normal plasma potassium level is dependent then on:
- Adequate dietary intake of potassium.
- Normal renal function.
- Normal gastrointestinal tract function.
- Normal production of aldosterone by adrenal glands.
- Maintenance of normal acid-base balance.
- Normal action of the sodium–potassium pump.
Laboratory measurement of sodium and potassium
Patient preparation
No particular patient preparation is necessary. Sodium and potassium are frequently requested on patients receiving fluids intravenously. Sampling blood from an arm into which IV fluid is being administered may cause errors and should be avoided.
Timing of sample
Blood may be sampled at any time for sodium and potassium but as these estimations may be made more than once a day on the same patient, it is important to record the time the sample was collected.
Sample requirements
Around 3–5 ml of venous blood is required. Measurement may be made on the serum or plasma recovered from a blood sample. If local policy is to use serum then blood must be collected into a collection tube without anticoagulant. Plasma estimation requires collection into a tube containing the anticoagulant lithium heparin. No other anticoagulant is suitable for sodium and potassium estimation.
Poor technique during blood collection may cause damage to the membrane of blood cells (haemolysis), resulting in erroneously high potassium concentration. This is because potassium is present at such high concentration within cells compared with plasma (Figure 4.2). Damage to (red and white) blood cells, that is haemolysis, results in a massive influx of potassium (and other cell contents) from cells into plasma. Haemolysis has relatively little effect on plasma/serum sodium concentration because its concentration in cells is low, compared with that of plasma. Haemoglobin, released from haemolysed red cells, changes the colour of plasma/serum from straw coloured to red, providing laboratory staff with a means of identifying haemolysed samples. Haemolysis may occur if blood is forced at pressure through syringe needles, if blood is shaken violently or frozen. It is a common occurrence if there has been any difficulty in sampling blood. Haemolysed samples are unsuitable for potassium analysis, although some modern analysers have the capacity to measure the degree of haemolysis, allowing a rough estimate of the ‘true’ plasma potassium concentration of haemolysed samples.
Some sodium and potassium analysers including those sited in intensive care units or recovery rooms allow immediate analysis on whole blood without the need to separate serum or plasma. Blood must be collected into a syringe or bottle containing the anticoagulant lithium heparin. There is no way of knowing if such samples are haemolysed.
Effects of storage
When blood is removed from the body, the available energy source, glucose required for normal maintenance of the sodium pump is soon used up and the sodium–potassium pump begins to fail. At this point potassium begins to leak from cells into plasma and sodium passes from plasma into cells. As with haemolysis, the effects are greater for potassium and erroneously high plasma/serum potassium levels are seen in samples left for more than a few hours. Blood for potassium estimation must be transported to the laboratory within an hour or so. Samples are best left at room temperature during any delay in transport. Blood left for more than three hours before transport to the laboratory is unsuitable for potassium analysis.
Interpretation of results
Reference range
Plasma/serum sodium | 133–146 mmol/L |
Serum potassium | 3.5–5.3 mmol/L |
(plasma potassium is slightly lower than serum potassium) | |
Critical values
Plasma/serum sodium | <120 mmol/L or >160 mmol/L |
Plasma/serum potassium | <2.5 mmol/L or >6.0 mmol/L |
Terms used in interpretation
Hyponatraemia | reduced plasma/serum sodium concentration, i.e. sodium <133 mmol/L |
Hypernatraemia | raised plasma/serum sodium concentration, i.e. sodium >146 mmol/L |
Hypokalaemia | reduced plasma/serum potassium concentration, i.e. potassium <3.5 mmol/L |
Hyperkalaemia | raised plasma/serum potassium concentration, i.e. potassium >5.3 mmol/L |
Causes of hyponatraemia
As we have seen sodium and water metabolism are inextricably linked. Plasma/serum sodium concentration is dependent on two variables; the amount of sodium in blood plasma (ECF) and the amount of water in blood plasma, that is (ECF) volume. Hyponatraemia, the most common electrolyte abnormality occurring in 15–30% of hospitalised patients1, may develop if sodium is lost from the body in excess of water (sodium depletion) or if there is an abnormal excess of water relative to sodium (sodium dilution). Hyponatraemia can also be classified according to clinical impression of the patients ECF volume status as: hypovolaemic hyponatraemia (volume depleted, water deficit); hypervolaemic hyponatraemia (increased volume, water excess); or euvolaemic hyponatraemia (normal volume, no significant water excess or deficit).
In general hyponatraemia is much more commonly due to disturbance of the mechanisms involved in maintaining water balance than in those involved in maintaining sodium balance. Hyponatraemia thus does not necessarily indicate sodium deficiency and can occur in those who have sufficient sodium and even an excess of sodium.
Sodium depletion
Abnormal losses of both sodium and water via the gastrointestinal tract occur during vomiting and diarrhoea, from skin during profuse sweating or as a result of burns. Haemorrhage represents a loss of both sodium and water. The effect that these losses have on serum sodium concentration depend on the sodium concentration of the fluid lost compared with that of plasma. Excessive vomiting, diarrhoea or sweating are generally associated with predominant water deficiency and therefore a tendency for plasma/serum sodium to rise. However, if these conditions are treated with fluid relatively deficient of sodium (i.e. hypotonic solutions), hyponatraemia may develop. This is a common cause of hyponatraemia.
Fluid lost during extensive burns and during haemorrhage has the same concentration as that of serum and in these cases, despite being sodium depleted, patients have a near normal sodium concentration. However, hyponatraemia may again develop if the fluid used to replace losses is, relatively to plasma, deficient of sodium (hypotonic).
Excessive loss of sodium in urine is the cause of the hyponatraemia that often accompanies diuretic therapy and contributes to the hyponatraemia seen in renal failure.
The hormone aldosterone regulates sodium loss in urine. A deficiency of the hormone results in increased inappropriate losses of sodium in urine. Addison’s disease is characterised by destruction of the adrenal gland; the resulting deficiency of aldosterone causes hyponatraemia.
Sodium dilution
Mild hyponatraemia is a frequent finding among any population suffering disease (the mean sodium concentration of hospital in-patients is 3–5 mmol lower than a control population in good health). The sick cell syndrome is thought to be the cause of this non-specific mild hyponatraemia frequently seen in generalised illness. The sick cell is one in which reduced cellular energy causes an abnormal increase in cell membrane permeability. This allows a slight abnormal shift of water from the ICF to the ECF, effectively diluting plasma sodium. This transient slight fall in serum sodium concentration usually requires no treatment and resolves as the underlying illness is treated.
Untreated diabetes mellitus is often associated with hyponatraemia. Glucose is an osmotically active substance. If, as is the case in diabetes, blood glucose concentration increases, then plasma osmolarity too increases. Water passes from cells to plasma to correct osmolality, thereby diluting sodium. This tendency to hyponatraemia in diabetes is compounded by the sodium loss in urine which accompanies the osmotic diuresis, as excess glucose is excreted in urine.
The importance of the antidiuretic hormone, AVP for renal regulation of water loss has already been discussed. The syndrome of inappropriate antidiuretic hormone (SIADH) results in abnormal retention of water, with dilution of sodium and therefore hyponatraemia. SIADH may complicate the course of many serious pathologies, including some lung cancers, infectious disease of the lungs, head injury, brain tumours and Guillain-Barré syndrome. Some drugs can also precipitate SIADH.
Water and sodium excess is a feature of oedema, accumulation of fluid in the interstitial space. Such fluid accumulation occurs in liver disease (cirrhosis), cardiac failure and chronic kidney disease. Despite increased total body sodium, water excess predominates in oedema with resulting hyponatraemia. Sodium replacement therapy is of course not indicated for this subset of hyponatraemic patients who have more than sufficient sodium but are retaining abnormal amounts of water. The causes of hyponatraemia are summarised in Table 4.1.
Causes of hypernatraemia
Hypernatraemia is much less common than hyponatraemia. Although excess sodium can cause hypernatraemia, relative water depletion is more often the cause.
Excess sodium
Hypernatraemia may occur during over vigorous sodium replacement therapy among patients with sodium depletion.
Uncontrolled secretion of aldosterone by a tumour of the adrenal gland is the cause of sodium retention of Conn’s syndrome (primary hyperaldosteronism). The kidneys respond to high plasma sodium by excreting less water; this tends to restore plasma sodium concentration towards normal but is usually insufficient and slight hypernatraemia is a common finding in Conn’s syndrome.
Excess cortisol (another hormone which affects renal loss of sodium), a feature of Cushing’s syndrome, has a similar effect.
Table 4.1 Causes of abnormal plasma/serum sodium and potassium.
| Causes of reduced plasma/serum sodium (hyponatraemia) |
|
| Causes of increased plasma/serum sodium (hypernatraemia) |
|
| Causes of reduced plasma/serum potassium (hypokalaemia) |
|
| Causes of increased plasma/serum potassium (hyperkalaemia) |
|
Water deficit
Despite maintenance of normal amounts of sodium, hypernatraemia will develop if water output in urine, sweat, faeces and expired air exceeds water intake. Inability of the kidneys to retain water may cause hypernatraemia in chronic kidney disease. Abnormal loss of fluid of relatively low sodium concentration is a feature of protracted vomiting, diarrhoea and sweating. All result in hypernatraemia if fluid intake is not increased to replace losses.
The thirst response is essential for adequate water intake. A minimum loss of water from the body each day is inevitable. Under normal circumstances the thirst response ensures that we take sufficient water to replace these losses. Unconscious patients and those who have sustained head injury involving damage to hypothalamic thirst centres within the brain are at increased risk of water depletion and therefore hypernatramaemia, because they have no effective thirst response.
The ability of the kidneys to conserve water when necessary by excreting urine of low volume and high concentration is dependent on adequate amounts of AVP (see Figure 4.4). A deficiency of AVP, or in some cases lack of AVP effect on the kidney tubules, is the cause of diabetes inspidus. This syndrome results in water depletion as urine of inappropriately high volume and low concentration is excreted. Failure of the pituitary to secrete AVP can be due to damage to the hypothalamus or the pituitary (e.g. head injury, neurosurgery). Some rare invasive tumours of the hypothalamus and pituitary can cause diabetes insipidus. Infections of the central nervous system (meningitis and encephalitis) sometimes precipitate diabetes insipidus.
In some types of diabetes insipidus, AVP production and secretion are normal, but the kidney tubules are unable to respond normally. This so called nephrogenic diabetes insipidus can be inherited, or precipitated by the action of some drugs (lithium, used in the treatment of manic depressive disorders is the most widely documented).
The principle causes of hypernatraemia are summarised in Table 4.1.
Causes of hypokalaemia
Although hypokalaemia can be a feature of chronic starvation, for example, in anorexia nervosa, inadequate potassium intake is a rare cause of hypokalaemia. Most cases are the result of increased losses from the body. Increased loss of potassium in urine is an unwanted side effect of some diuretic drugs (e.g. frusemide, Lasix). Diuretic therapy is probably the most common cause of hypokalaemia. Potassium supplements may need to be prescribed for patients receiving diuretic therapy.
The adrenal cortex hormone aldosterone regulates potassium excretion in urine. Excess aldosterone causes abnormally high urinary losses of potassium with resulting hypokalaemia and is a feature of Conn’s syndrome, in which there is excessive aldosterone secretion by an adrenal tumour. Raised levels of aldosterone in part accounts for the severe hypokalaemia that occurs in those with the rare inherited condition Bartter’s syndrome, and in a similar condition precipitated by liquorice abuse!
Like sodium, potassium can be lost in abnormally high amounts from the gastrointestinal tract; for example, severe acute diarrhoea and the chronic diarrhoea associated with purgative (laxative) abuse can result in sufficient potassium to be lost from the body to cause hypokalaemia. Vomiting is not usually associated with significant potassium depletion except in the specific case of pyloric stenosis, in which the projectile vomiting of acid contents of the stomach causes alkalosis.
Hypokalaemia may be caused not by loss of potassium from the body but by a shift of potassium from the ECF into cells. Such an abnormal shift occurs for one of two reasons: increased activity of the sodium–potassium pump or if there is a hydrogen ion deficit, that is raised blood pH (alkalosis). In the first case, potassium passes into cells in exchange for sodium; in the second, potassium passes into cells in exchange for hydrogen ions (to correct ECF pH). The pancreatic hormone insulin increases the activity of the sodium–potassium pump, so that a shift of potassium from ECF to cells occurs during insulin therapy for diabetic ketoacidosis. This contributes to the hypokalaemia that often occurs as diabetic ketoacidosis is treated. Incidentally this action of insulin is used therapeutically to reduce plasma potassium in those with severe hyperkalaemia, whatever the cause.
The passage of potassium from ECF to cells in exchange for hydrogen ions is a feature of alkalosis and is the reason for the hypokalaemia of pyloric stenosis. This tendency to hypokalaemia during alkalosis is potentiated by increased renal excretion of potassium as hydrogen ions are conserved to in an attempt to raise blood pH. Other causes of alkalosis that may be associated with hypokalaemia are considered in Chapter 7.
The causes of hypokalaemia are summarised in Table 4.1.
Causes of hyperkalaemia
Excessive intake of potassium during treatment with potassium supplements to correct potassium depletion may cause hyperkalaemia but is very rarely the sole cause. Reduced renal excretion of potassium is the more common mechanism. As kidneys fail they lose the ability to excrete potassium in urine; chronic kidney disease is the most common cause of hyperkalaemia and hyperkalaemia is usually a feature of acute kidney injury.
Autoimmune destruction of the adrenal glands, that is Addison’s disease, results in a deficiency of aldosterone, the hormone which regulates renal excretion of potassium. The hormone deficiency results in reduced potassium excretion and therefore hyperkalaemia.
Most of the human body’s potassium is contained within cells; widespread damage to cells results in release of potassium into the ECF. For example, severe trauma may result in hyperkalaemia, as may the massive cell destruction associated with cytotoxic therapy for the treatment of leukaemia and the widespread skeletal muscle cell destruction that characterises the usually trauma-related syndrome rhabdomyolysis. This tendency to hyperkalaemia due to tissue destruction will be potentiated by any degree of renal dysfunction.
Potassium passes from cells into the ECF in exchange for hydrogen ions if blood is abnormally acidic. For this reason acidosis is often associated with hyperkalaemia. The causes of acidosis are outlined in Chapter 7. Special mention however is made here of the acidosis associated with untreated diabetes.
Untreated diabetic ketoacidosis is usually associated with hyperkalaemia although whole body potassium is depleted. Potassium depletion occurs due to increased urinary losses of potassium during the osmotic diuresis caused by urinary excretion of glucose. This potassium depletion is masked however by movement of potassium out of cells into the ECF, due to acidosis, and dehydration consequent on the massive amounts of water lost during the osmotic diuresis. Although severely depleted of potassium, serum levels are normal or high. The potassium depletion soon becomes apparent as the acidosis and dehydration are corrected; hypokalaemia develops as potassium returns to cells and rehydration is effected.
It is as well to emphasise the possibility that a raised potassium level might be due solely or partially to poor practice during collection, storage and transport of specimens (see sample collection).
The causes of hyperkalaemia are summarised in Table 4.1.
Consequence of abnormal plasma/serum sodium potassium concentration
Signs and symptoms of hyponatraemia
The clinical effect of low serum sodium depends on the cause, the magnitude of the abnormality and rapidity of onset. Mild hyponatraemia (130–133 mmol/L) is not usually associated with symptoms, but most patients with a plasma sodium of less than 125 mmol/L will experience some symptoms; these will be more severe if the decrease is rapid (within 24–48 hours). As we have seen, most cases of severe hyponatraemia are due to relative water excess. Symptoms result from over hydration of cells; the cells of the brain are particularly sensitive to this water excess and neurologic symptoms predominate. Headache, lethargy, mental depression and confusion may develop. Severe hyponatraemia (plasma sodium <115 mmol/L), particularly of rapid onset, is associated with convulsions and coma; if left untreated severe hyponatraemia can be fatal.
If hyponatraemia is accompanied by significant water depletion, as in say late Addison’s disease, symptoms of low ECF volume (circulatory shock) predominate; these include reduced blood pressure, tachycardia and dizziness. If, on the other hand, hyponatraemia is accompanied by significant water excess, symptoms associated with increased ECF volume predominate; these include weight gain, oedema, hypertension and breathlesness on exertion (pulmonary oedema).
Signs and symptoms of hypernatraemia
Most cases of hypernatraemia result from a water deficit of both the ECF and ICF. Rapidity of changes increases the severity of symptoms, which are essentially those of dehydration: thirst, dry mouth, difficulty in swallowing and red swollen tongue. Cerebral cell dehydration causes neurological symptoms including confusion and lethargy, increased neuromuscular activity (twitching) and eventually coma. Like hyponatraemia, severe hypernatraemia can be fatal.
Signs and symptoms of hypokalaemia
Symptoms of hypokalaemia do not usually arise until potassium concentration falls below 3.0 mmol/L. But when they do arise symptoms are related to the function of potassium in transmission of nerve impulses to muscle.
Muscular weakness associated with general lethargy is the most common symptom. Constipation due to impaired muscle tone of the gastrointestinal tract may be a problem. Muscular paralysis may occur in patients with severe hypokalaemia (plasma potassium <2.5 mmol/L).
Cardiac muscle is frequently affected resulting in cardiac arrhythmias including tachycardia and sinus bradycardia. Severe hypokalaemia can result in ventricular fibrillation and cardiac arrest. Electrocardiographic (ECG) examination is frequently indicated for patients with hypokalaemia; typical ECG changes include: prominent U waves; decreased amplitude and broadening of T waves; and S-T segment depression. The toxic effects of digoxin therapy are potentiated by hypokalaemia. Metabolic alkalosis (discussed in Chapter 7), which can accompany hypokalaemia, may result in symptoms of tetany.
Signs and symptoms of hyperkalaemia
Hyperkalaemia may be accompanied by vague feelings of muscle weakness, not as pronounced as those that are characteristic of hypokalaemia. Affected patients may be apathetic or even confused. Slurred speech is occasionally evident. The most significant effect of hyperkalaemia however is life-threatening changes in cardiac muscle contraction (cardiac arrhythmias). As serum potassium rises above 7.0 mmol/L there is a real risk of cardiac arrest and sudden death. Electrocardiograph (ECG) examination is indicated for all patients with hyperkalaemia; characteristic ECG changes associated with hyperkalaemia include: tall, peaked T waves; low or missing P waves; and broadening of the QRS complex. Urgent potassium lowering therapy is indicated for patients with moderate/severe hyperkalaemia.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


