CHAPTER 32. Shock Emergencies
Reneé Semonin Holleran
Shock is a clinical manifestation of inadequate tissue perfusion. It is a life-threatening, generalized maldistribution of blood flow that results in failure of the delivery and utilization of oxygen. There are multiple causes of shock; however, the pathophysiology of shock is generally the same. It is important to remember that vital sign changes do not define shock. Evidence of inadequate tissue perfusion based on physical examination and suspicion based on history are the best methods of identifying shock. 1. and 12.
The shock state is also associated with systemic inflammatory response syndrome (SIRS). The causes of shock, such as an infection or volume loss, serve as a primary insult, or the body’s response to hemorrhage or infection as a secondary response, that triggers SIRS. Inflammatory markers can be detected in the blood of all patients in shock. However, biomarker levels and the significance of these levels vary depending on the cause of the shock state. 1
This chapter will discuss the pathophysiology of shock, the categories of shock, shock management in the emergency department (ED), and the effects of shock on selected patient populations.
PATHOPHYSIOLOGY
Shock results from inadequate tissue perfusion that leads to failure of the delivery and/or use of oxygen, causing tissue dysoxia. 1 In the presence of hypoxia and inadequate tissue perfusion, cells do not receive oxygen and nutrients (especially glucose), nor are they able to remove waste products. Cellular damage and eventual death ensues when cellular oxygen demands exceed the tissue’s oxygen supply.
Normal cell metabolism requires an aerobic environment to break down glucose and oxidize substrates. Enzyme-mediated chemical reactions transfer energy from this process into adenosine triphosphate (ATP). Oxidative energy synthesis of ATP is necessary for cell survival and is a fundamental characteristic of life. This process is also referred to as cellular respiration.
Cellular respiration occurs within organelles known as mitochondria, which are located in the cell’s cytoplasm. Mitochondria are the site of ATP synthesis and energy production. Lysosomes in the cytoplasm store hydrolytic or digestive enzymes that mediate chemical reactions within the cell.
The production of cellular energy and synthesis of ATP is dependent on a continuous oxygen supply. Availability of oxygen is influenced by blood flow, oxygen saturation, and cardiac output. Oxygen consumption (VO 2) is the amount of oxygen removed by the tissues for metabolism. Oxygen debt refers to the difference between cellular demand for oxygen and cellular consumption of available oxygen. A continuous oxygen debt related to tissue hypoxia creates an anaerobic environment that adversely affects cellular metabolism. Oxygen delivery (DO 2) depends on the oxygen content of arterial blood and the cardiac output.
Anaerobic metabolism leads to the accumulation of lactate, hydrogen ions, and inorganic phosphates within the cell. ATP production is diminished, and protein production is compromised, which results in damage to the mitochondria of the cell. Damage to the mitochondria triggers problems with electron transport and the activation of apoptosis (programmed cellular destruction). Intracellular enzymes that are activated by these changes release enzymes that further deplete ATP stores (cellular energy) and damage cellular membranes, including noninjured cells.
A shift of sodium into the cell to increase cellular fluid causes the displacement of potassium outside of the cell. When cells swell from the absorption of interstitial fluid, this swelling narrows the capillary lumens and further decreases the supply of oxygen and nutrients to the cells. Changes in potassium interfere with nervous, cardiovascular, and muscular cell function. Eventually, energy-dependent potassium channels fail, causing arterioles to dilate and lead finally to uncompensated shock.
The cause of shock serves as a trigger to initiate a systemic inflammatory response. Whether it is direct tissue damage, for example, a brain contusion or liver laceration, or an infection from burn wounds, the body initiates an immune response that releases proinflammatory cytokines and phospholipids. This sets off a cascade of changes that assist in protecting the body. Proinflammatory cytokines activate cells that function within the immune system. This results in a hyperinflammatory response (i.e., SIRS). One function of the immune system is to stop the programmed cell death (apoptosis) that occurs with SIRS. If the cause of shock is not recognized and managed early, SIRS will continue and result in the production of arachidonic acids, prostaglandins, thromboxanes, and other metabolites that will continue the inflammatory response. 10
In an effort to maintain homeostasis, antiinflammatory mediators are produced. Unfortunately, these mediators are responsible for immunosuppression that leaves the patient at risk for further infection and/or septic complications. The body’s ability to successfully balance proinflammatory and antiinflammatory responses so that it can heal or fight off an infection depends on early and appropriate interventions to manage and treat the cause of the shock state.
The activation of cytokines, arachidonic acid metabolites, and other toxins instigates the plasmatic cascade system, which plays a role in endothelial and parenchymal cell death and in coagulation. This can result in an inability to heal, as well as development of disseminated intravascular coagulation (DIC), 10 and becomes a vicious cycle that must be stopped as early as possible.
Glucose metabolism is also impaired. The stress of the shock state triggers gluconeogenesis to provide fuel for system functioning and healing. In addition, the liver and kidneys produce more glucose in response to the secretion of epinephrine, norepinephrine, glucagon, and cortisol as part of the body’s stress response. Even though the body produces more glucose as a response to stress, insulin resistance occurs because of increased production of serum cytokines. 20 Increased production of glucose and insulin resistance actually impair cellular growth and metabolism. This has lead to the development of protocols to use insulin therapy in the care of patients in shock, particularly sepsis. 19. and 20.
Figure 32-1 provides a summary of impaired cellular metabolism that occurs as a result of inadequate tissue perfusion and impaired oxygen and glucose use.
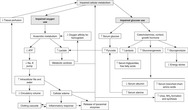 |
| FIGURE 32-1 Impairment of cellular metabolism by shock. ATP, Adenosine triphosphate. (From Baldwin KM, Morris SE: Shock, multiple organ dysfunction syndrome, and burns in adults. In McCance KL, Huether SE: Pathophysiology: the biologic basis for disease in adults and children, ed 5, St. Louis, 2006, Mosby.) |
Stages of Shock
Regardless of the cause of the shock state, the body mobilizes a series of responses to compensate. Compensatory responses are stimulated by decreasing tissue perfusion. Ideally the outcome of these compensatory mechanisms is restoration of cardiac output and tissue perfusion. To accomplish this, blood is shunted from the kidneys, gastrointestinal tract, liver, and skin to vital organs (the heart, lungs, and brain). Key compensatory responses include the baroreceptors, sympathetic nervous system, fluid shifts, and endocrine system. Without these compensatory mechanisms, shock progresses and ultimately death occurs. Table 32- 1 summarizes the progression of the shock state.
| Stage | Pathophysiology |
|---|---|
| Compensated (nonprogressive) stage | Various receptors sense the drop in systemic pressure and initiate a cascade of physiologic changes. Homeostatic compensatory mechanisms, mediated by the sympathetic nervous system, increase end organ perfusion. Aerobic metabolism changes to anaerobic metabolism and lactic acid is produced. |
| Uncompensated (progressive) stage | Compensatory mechanisms begin to fail. Mechanisms initially helpful, now cause more injury. Cellular derangement occurs which causes organ death. Severe lactic acidosis and inflammatory/immune system activation contribute to this. |
| Irreversible (refractory) stage | In refractory shock, no treatment can reverse the shock process. The cause of the shock is indistinguishable. The clinical progression of the shock state is unique to the patient and is influenced by factors such as the cause of the shock, the age of the patient, severity and duration of the hypoperfused state, and presence of preexisting diseases. |
Compensatory Mechanisms
When the patient “goes into shock,” there are multiple compensatory mechanisms that are triggered within the body in an attempt to compensate and maintain a state of homeostasis. The following is a discussion of these.
Baroreceptors
Baroreceptors are a collection of specialized neural tissues located in the aortic arch and bifurcation of the common carotid arteries. Baroreceptor reflexes respond to small changes in vascular tone or pressure. Inhibition of baroreceptors by the vasomotor center of the brain in response to decreasing cardiac output results in sympathetic stimulation followed by peripheral vasoconstriction in an effort to increase circulating volume and maintain blood pressure. Similarly, the resulting decrease in vagal tone decreases coronary resistance in an attempt to improve myocardial oxygen supply. 12
Sympathetic Nervous System
Stimulation of the sympathetic nervous system by decreases in circulating volume and cardiac output results in release of epinephrine, norepinephrine, and other catecholamines that stimulate α- and β-receptors. Stimulation of α-receptors is followed by arteriolar and venous vasoconstriction, which shunts blood to organs. Other effects include stimulation of the adrenal glands, which ultimately leads to fluid retention by the kidneys. The chronotropic effect is tachycardia. β-Receptor stimulation has a positive inotropic effect, increasing myocardial contractility and improving coronary artery blood flow. The α- and β-adrenergic effects augment venous return, increasing ventricular filling or preload, heart rate, and myocardial contractility, which facilitates ventricular emptying and improves cardiac output and blood pressure. 6
Shift of Interstitial Fluid
Normal distribution of body fluid is 75% intracellular and 25% extracellular. Of the extracellular fluid, one third is located intravascularly; the remainder is interstitial. Hydrostatic pressure and plasma colloid oncotic pressure (COP) are forces that maintain normal fluid distribution between the intravascular and interstitial compartments. Hydrostatic pressure pushes fluid from the arterial end of the capillary bed into the interstitial space. COP pulls fluid into the venous capillary bed because of plasma proteins such as albumin.
Endocrine Response
The endocrine system response to decreased circulating volume or decreased cardiac output is a result of the interaction of the adrenal glands, the kidneys, the pituitary gland, and the lungs. Components are the renin-angiotensin-aldosterone system and antidiuretic hormone (ADH).
RENIN-ANGIOTENSIN-ALDOSTERONE SYSTEM
Renal hypoperfusion and mediation of β-receptors by the sympathetic nervous system secondary to shock causes release of renin. Renin activates conversion of angiotensinogen to angiotensin I, which is then converted by the lungs into angiotensin II and causes release of aldosterone and produces vasoconstriction. Aldosterone promotes sodium reabsorption within the renal tubule. Water follows reabsorption of sodium, so the net effect is water movement from the interstitial space into the intravascular space. Ideally, circulating volume and cardiac output increase. There is a corresponding decrease in urinary output as a result of this response.
ANTIDIURETIC HORMONE.
ADH is released by the posterior pituitary gland in response to increased plasma osmolarity, altered circulating volume, and in response to angiotensin. ADH causes sodium and water reabsorption from the distal renal tubules in an attempt to increase circulating volume and cardiac output. Urine output decreases, and urine specific gravity increases as a result of this process.
Adrenal Gland Response
The adrenal glands are stimulated by the sympathetic nervous system when a patient is in shock. This causes the release of epinephrine and norepinephrine as part of the stress response. The release of these catecholamines causes tachycardia and initially improves cardiac output to focus on end-organ perfusion. Both epinephrine and norepinephrine cause peripheral vasoconstriction to increase peripheral vascular resistance and increase blood return to the heart.
The triggering of the stress response stimulates the hypothalamus to secrete corticotropin-releasing hormone, which stimulates the pituitary to release adrenocorticotropic hormone (ACTH). This causes the release of cortisol (another stress-response hormone) from the adrenal glands. Cortisol will elevate blood glucose and renal retention of water and sodium. 6
CATEGORIES OF SHOCK
Shock has been categorized or classified in a number of ways. The one most commonly used is based on the cause of shock, for example, hemorrhagic because of blood loss. Shock may also be described by type, including hypovolemic, cardiogenic, distributive, and obstructive. Finally, another classification looks at alterations in circulating volume, alterations in the capability of the heart to move the circulating volume, and alterations in peripheral vascular resistance. The following is a description of the categories of shock based on the cause.
Hypovolemic Shock
Hypovolemic shock results from loss or redistribution of blood, plasma, or other body fluids, which ultimately leads to overall reduction in intravascular volume. Hypovolemic shock is primarily an alteration in circulating volume. Hemorrhagic shock is a form of hypovolemic shock. Hypovolemic shock is the type of shock seen most frequently by the emergency nurse. It is a direct result of reduction in intravascular volume. Decreased circulating volume can be caused by loss or redistribution of whole blood, plasma, or other body fluids. Loss of volume in the intravascular space causes a reduction in preload. Preload describes the force that stretches the cardiac ventricles to an initial length. The resting force on the heart is determined by the pressure in ventricles at the end of diastole. When blood or volume is lost, there is inadequate fluid returning to the heart and that has an impact on preload. Redistribution, or third-space sequestration, occurs when fluid shifts from the intravascular compartment to the interstitial space. This can occur with changes in capillary permeability or capillary fluid pressures, as in burn injuries or sepsis, and can also have an impact on preload. Actual volume loss is associated with traumatic injury, posterior nasal bleeds, intraabdominal hemorrhage, significant vaginal bleeding, gastrointestinal bleeding, or excessive vomiting and diarrhea. Table 32-2 illustrates the physiologic response to hemorrhage based on a 70-kg male.
| Class % Blood Loss | Pulse | Blood Pressure | Pulse Pressure | Level of Consciousness | Respiratory Rate | Urinary Output |
|---|---|---|---|---|---|---|
| Class One (I) Up to 15% (up to 750 ml) | <100 | Normal | Normal or increased | Slightly anxious | 14-20 | >30 ml/hr |
| Class Two (II) 15%-30% (750-1500 ml) | >100 | Normal | Decreased | Mildly anxious | 20-30 | 20-30 ml/hr |
| Class Three (III) 30%-40% (1500-2000 ml) | >120 | Decreased | Decreased | Anxious, confused | 30-40 | 5-15 ml/hr |
| Class Four (IV) >40% (>2000 ml) | >140 | Decreased | Decreased | Anxious, confused | 30-40 | 5-15 ml/hr |
Cardiogenic Shock
Cardiogenic shock occurs when the heart fails as a pump, causing a significant reduction in ventricular effectiveness. Cardiac output decreases and tissue perfusion diminishes, while left ventricular end-diastolic pressure increases. Injury to the myocardium impairs contractility, which decreases ventricular emptying. Cardiogenic shock continues to carry a high mortality rate; most patients die within 24 hours; others may live only a few days. Cardiogenic shock is caused by myocardial infarction with damage to greater than 40% of the left ventricle, severe myocardial contusion, valvular heart disease, cardiomyopathies, ruptured papillary muscle, ruptured ventricular septum, and dysrhythmias such as third-degree heart block. Myocardial damage may occur as the result of a single infarction or subsequent to multiple events.
When pump failure occurs, the myocardium cannot forcibly eject blood. Stroke volume decreases because of decreased contractility, which decreases cardiac output and blood pressure. Subsequent alterations in tissue perfusion precipitate myocardial ischemia and extend the region of injury, further compromising cardiac contractility. Myocardial contractility is also affected by hypoxemia, metabolic acidosis, ventricular diastolic volume, and sympathetic nervous system stimulation. Poor myocardial contractility causes inadequate emptying of the left ventricle.
Incomplete emptying of the left ventricle during diastole elevates pressures in the left ventricle, left atrium, and pulmonary vessels. There is a corresponding increase in pulmonary pressures as pulmonary capillaries leak fluid into the alveolar spaces, causing pulmonary edema. Elevated pulmonary pressures increase right ventricular and atrial pressures, which contribute to right-sided heart failure.
Increased myocardial performance as a compensatory response to shock translates to increased myocardial oxygen demand and increased myocardial ischemia, which further compromises cardiac output and eventually progresses to cardiovascular collapse.
Neurogenic Shock
Neurogenic shock is most often associated with acute spinal cord disruption from trauma or spinal anesthesia. Other causes of neurogenic shock are brain injury, hypoxia, depressant drug actions, and hypoglycemia associated with insulin shock. In neurogenic shock, outflow from the vasomotor center in the medulla is inhibited or depressed, causing loss of sympathetic vasomotor regulation. Uncontested parasympathetic responses cause vasodilatation and loss of sympathetic tone. Inhibition of sympathetic innervation impedes release of norepinephrine and interferes with the body’s ability to vasoconstrict. Consequently, venous return and cardiac output decrease.
Septic Shock
Septic shock is defined as the presence of sepsis with refractory hypotension. 14 Septic shock is caused by an infectious agent or infection-induced mediators. The body responds through both hyperinflammatory and antiinflammatory means. Endotoxins released by the invading organisms prompt release of hydrolytic enzymes from weakened cell lysosomes, which causes cellular destruction of bacteria and normal cells. When the body is unable to control the proinflammatory mediators, it produces a systemic inflammatory response. As a result, there is widespread cellular dysfunction that brings about acute respiratory distress syndrome (ARDS), DIC, multiple organ dysfunction syndrome (MODS), and eventually death. The mortality rate fromseptic shock is 50% and increases when patients have comorbid problems such as immunosuppression, diabetes, or cardiovascular disease.
Anaphylactic Shock
Anaphylaxis is an acute immune and inflammatory response to an allergen. An allergen is an antigen to which someone has become sensitized, for example, insect venom, pollens, shellfish, and antibiotics. Anaphylactic shock is a profound hypersensitivity reaction with a systemic antigen-antibody response. Antibodies are formed on initial exposure to a foreign protein. Current research suggests that the pathophysiology of anaphylactic shock results from a profound reduction in venous tone and extravasation of fluid, which causes reduced venous return and depressed myocardial function. 3 Clinical manifestations are usually acute and sudden and include hypotension, altered mental status, and in some cases cardiac arrest. The release of histamine causes peripheral skin flushing, hypotension, and tachycardia. The initial clinical manifestations of anaphylactic shock may include anxiety, shortness of breath, nausea, vomiting and diarrhea, hives, urticaria, and sensations of burning and itching of the skin. 12
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Get Clinical Tree app for offline access
