As part of their early routine health assessment, all babies born in the UK are, with parental agreement, submitted for the blood tests that are the subject of this chapter. So of all laboratory blood tests, these are the most universally applied. The tests, collectively known as the newborn bloodspot screening test (formerly called the Guthrie test), require a single heel-prick blood sample collected, usually by the midwife or health care visitor, when babies are five to eight days old. The blood is collected directly onto a newborn bloodspot card (a special filter paper, formerly called a Guthrie card), which is sent by post to one of 20 specialist laboratories located throughout the UK. The object of this nationally co-ordinated blood testing is to identify those babies who are at high risk of suffering any one of five rare but serious congenital conditions that have lifelong health significance. Early diagnosis and treatment reduces the severity of all five conditions and may be life saving. The five conditions are: phenylketonuria (PKU), congenital hypothyroidism (CH), cystic fibrosis (CF), medium-chain acyl CoA dehydrogenase deficiency (MCADD) and sickle cell disease (SCD).
In this chapter we consider each of these conditions in turn and the specific blood tests used to screen for them, but first there follows a brief general discussion about screening for disease and nationally co-ordinated screening programmes to place the one under discussion here in a wider context.
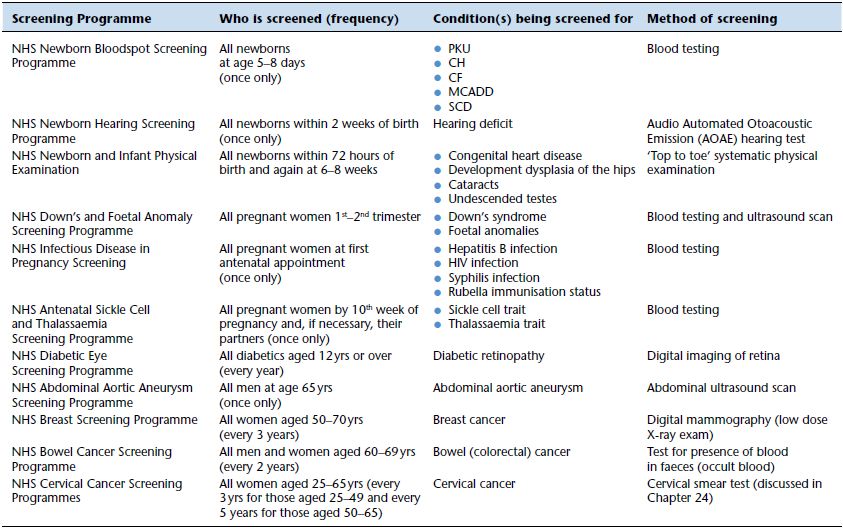
Table 23.1 UK Nationwide screening programmes.
Screening tests and national screening programmes
Screening tests are those that are offered to apparently healthy people in order to identify those who may be at high risk of a particular disease or condition. These tests are not intended to identify those with a particular disease or condition – that is the object of the diagnostic process, which is invariably more expensive and in one way or another less easily applied to large numbers of people. The value of the effective screening test is that it relatively easily and cheaply eliminates the vast majority who are not at risk of the disease or condition being screened for, so that only a very small number need be submitted for the usually more expensive, complex diagnostic testing and clinical assessment that either confirms or excludes the disease.
It is only considered justifiable to screen for a disease if early identification of that disease before symptoms develop leads to a better outcome. If there is no effective treatment or early treatment has no benefit there is arguably limited value in knowing you have the disease, indeed the knowledge may, on balance, have harmful effect.
The screening process has an ethical dimension because it exposes those who do not have the disease – the vast majority – to a process from which they gain no tangible health benefit. Indeed if such people screen positive they often have to undergo extra tests and investigations and be caused the anxiety associated with knowing they might have the disease, again without any benefit. For any proposed screening process the benefit gained by those who have the disease must be rigorously weighed against the potential harm that the screening process might cause those who do not have the disease.
Finally, no screening procedure is entirely reliable. For a number of reasons, some avoidable and some unavoidable, a small minority of those with the disease will be given a negative screen result, and a small minority of those without the disease will be given a positive screen result. Either scenario may have devastating consequence for the, thankfully few, individuals concerned and national screening programmes are constantly updated in the light of new research that reveals ways in which the number of false positive and false negative screening results can be minimised.
The limitations and cost of screening determine that national screening programmes are only used to screen for a small number of diseases/conditions, although it is true that the number has increased in recent years and the appropriateness of extending screening programmes is under constant review. Table 23.1 lists the 11 national screening programmes currently operational within the UK.
The screening process does not necessarily involve laboratory testing but clinical laboratories have a role in six of the 11 programmes, including two that are discussed in this book: the cervical cancer screening programme (Chapter 24) and the newborn bloodspot screening programme.
Newborn bloodspot programme – sample collection
As with all other UK nationally co-ordinated screening programmes, the effectiveness of the newborn bloodspot programme depends on the cooperative effort of several groups of healthcare workers, all working to well defined standards of quality and timeliness at each step of what is a quite complex process. The standards of the programme, for example, demand that laboratory staff complete all screening tests, including where necessary repeat analysis, within four days of receiving the bloodspot sample. On the same day that a positive screening result is identified, the defined local clinical liaison team (CLT) must be informed by telephone. On that day the CLT are required to arrange an urgent (ideally next day) follow up clinic appointment for diagnostic blood testing/clinical assessment of the baby, and parental counselling. Diagnosis should be confirmed within a few days of that appointment, using a clearly defined protocol. When all standards are observed, babies found to be affected by one of the conditions being screened for start appropriate treatment within two to four weeks of birth. Delay beyond this may have lifelong deleterious effect.

The first step in the process, usually the responsibility of midwife or health visitor, is collection of the bloodspot sample, after gaining informed consent of parents. The detail of standards for bloodspot sample collection is contained in published guidelines1 from which the following bullet points are derived:
- The bloodspot sample should be taken on day five, but in exceptional circumstances between days five and eight (day zero is taken as day of birth).
- Details must be entered on the bloodspot card (Figure 23.1) at the time of blood sampling. It is mandatory to include the baby’s identifying NHS number; the sample cannot be processed in the laboratory if this is absent or other details are undecipherable. Bar coded labels, that contain all essential details relating to the baby, are preferred.
- The baby’s heel from which blood is sampled must be clean and dry – it is essential that there is absolutely no trace of faecal matter because faeces contains large amounts of trypsin, a substance measured in blood to screen for cystic fibrosis. Failure to clean the heel sufficiently could lead to faecal contamination of the blood sample and a falsely positive screening result.
- An automated incision device that penetrates no more than 2 mm should be used to obtain the sample – manual lancets should not be used. The bottom (fleshy) surface of the heel should be punctured, not the back of the heel where the heel bone (calcaneus) can be felt.
- The aim is to fill each circle on the bloodspot card using a single drop of blood. Allow blood to form a drop on the heel. It is important that blood flows freely without the need to squeeze the foot. Allow one spot of blood to drop onto each of the circles. Do not allow the card to make contact with the heel. There should be sufficient blood for it to seep naturally through to the other side of the card and evenly saturate the whole of each circle. Insufficient sample, evident as multiple small spots of blood within the circle, will be rejected by the laboratory and a repeat requested. Too much blood caused by layering of blood drops on top of each other can cause erroneous results.
- Allow the bloodspots to air dry before placing in the glassine envelope and place the bloodspot card in the prepaid addressed envelope and post (first class) on the day the sample is collected. Standards demand that bloodspot cards are received by the laboratory within three days of sample collection.
- Blood transfusion prior to bloodspot sampling can adversely affect screening tests for SCD. This is of course only usually an issue for sick or premature babies admitted to neonatal care units. All these babies should have a bloodspot sample on admission in case they have a blood transfusion before day five. This is stored labelled as ‘pre-transfusion sample’ and must be sent with the normally timed sample if they have a blood transfusion in the interim. It can be discarded if no blood transfusion is given by day five. It is important to record any transfusion history on the bloodspot card.
Conditions screened for with the bloodspot test
Phenylketonuria (PKU)
This is an inherited disturbance of amino acid metabolism, specifically the amino acid phenylalanine 2. It affects around 1 in 10 000 babies; so, as the current annual UK birth rate is around 800 000, close to 80 PKU affected babies are born every year in the UK.
The principle biological significance of all amino acids (there are 23 in total) is as the building blocks of all proteins. Some amino acids, including phenylalanine, cannot be synthesised in the body and must be provided for in the diet; they are called the ‘essential’ amino acids. We obtain all the phenylalanine we need to synthesise proteins only by eating protein-containing foods. Tyrosine is an example of an amino acid that can be synthesised in the body; it is a ‘non-essential’ amino acid. In the body tyrosine is made from phenylalanine; the two are structurally very closely related.
The conversion of phenylalanine to tyrosine, which occurs principally in the cells of the liver, is a metabolic process that depends on the enzyme phenylalanine hydroxylase (PAH). Those with phenylketonuria have one of many possible inherited defects (mutations) in the gene that codes for synthesis of PAH. This results in absent or reduced activity of PAH (depending on the particular mutation), and therefore absent or reduced conversion of phenylalanine to tyrosine.
The condition is inherited in an autosomally recessive mode, meaning that it is necessary to inherit two copies of the mutated PAH gene, one from each parent. Those who inherit just one copy are unaffected ‘carriers’ of the condition. There is a one in four chance that a baby born to parents who are both healthy ‘carriers’ will be affected by PKU.
As a consequence of the inherited genetic defect, phenylalanine accumulates in blood and tissues. Some is metabolised to a group of substances known collectively as phenylketones, which are excreted in urine. The presence of phenylketones in urine is called phenylketonuria, which gives the condition its name, and once provided the only diagnostic signal of PKU. However, the most significant clinical consequence of PKU stems from the toxic effect of accumulating phenylalanine on brain function. Without early treatment very soon after birth, PKU in its most severe ‘classical’ form results in permanent brain damage and profound mental disability manifest as progressive intellectual impairment, accompanied by a range of symptoms that may include seizures and autism. Late in childhood or adolescence behavioural and psychiatric problems may emerge.
Since accumulating phenylalanine is the problem, and all phenylalanine is derived from food, treatment of PKU is based on simply restricting phenylalanine intake. Introduction of a phenylalanine-restricted diet soon after birth prevents brain damage and nearly all of the PKU associated neuro-psychological problems. The diet, which is essentially a very low protein diet almost devoid of phenyalanine, must be continued certainly until adolescence and ideally for life; efficacy must be monitored by regular blood phenylalanine measurement.
The PKU screening test
Screening for PKU is based on measurement of phenylalanine (and if necessary tyrosine) concentration in the blood spot sample by an automated technique known as tandem mass spectrometry. This technique allows phenylalanine/tyrosine measurement in a few minutes and the ability to process up to 600 bloodspot samples every day. The technique is suited for simultaneous measurement of all amino acids and many intermediary metabolites from a single sample, and so is a potential screening tool for many other congenital metabolic defects, including MCADD (explained further).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


