- Defining haemostasis
- Platelets and their role in haemostasis
- The blood clotting cascade – its role in haemostasis
- Fibrinolysis and D-dimer production
- What is being measured in the prothrombin time test
- Causes/consequences of increased platelet count
- Causes/consequences of decreased platelet count
- Venous thromboembolism (VTE) and its prevention
- Role of D-dimer test in diagnosis of VTE
Blood loss following blood vessel injury is minimised by the capacity of blood to form a physical plug at the site of the vessel injury. The complex physiological process that ensures this life preserving property of blood is called haemostasis. Blood must retain its ability to quickly form a physical plug of coagulated blood at the site of vessel injury, preventing undue blood loss, whilst remaining fluid and free of coagulated blood within undamaged vessels. Disturbance of this fine balance, a feature of many disease processes, can result in either an increased tendency to bleed if the coaguablity of blood is less than normal, or formation of a blood clot (thrombus) within patent undamaged blood vessels, if coagulability is abnormally increased.
Four of the five blood tests discussed in this chapter, platelet count, prothrombin time (PT), activated partial thromboplastin time (APPT) and thromboplastin time (TT) are primarily used in the first line investigation of patients who are suspected of suffering disease associated with an increased tendency to bleed, whilst the D-dimer test is used in the assessment of patients suspected of suffering thrombotic disease, that is disease associated with increased tendency of blood to coagulate and form thrombi inappropriately within patent blood vessels.
Patients suffering, or at high risk of suffering, thrombotic disease are treated with anticoagulant drugs that artificially decrease the coagulability of blood. Two of the blood tests considered in this chapter, prothrombin time (PT) and activated partial thromboplastin time (APPT) are routinely used to monitor this anticoagulant therapy.
Normal physiology
The sequence of events that leads to formation of a stable fibrin plug and cessation of bleeding (haemostasis) following blood vessel injury is described in Figure 17.1.
The first physiological response to blood vessel injury is local vasoconstriction, which reduces blood flow to the injured site thereby minimizing to some extent blood loss. Vessel injury also initiates two further physiological responses. The first is platelet adhesion and aggregation with formation of a physical plug of platelets at the site of vessel injury, and the second is initiation of the so called clotting cascade which results in production of the structural protein, fibrin. Fibrin strands join together (polymerise) and the fibrin polymers cross-link. The effect is a growing linked mesh of fibrin forming around and between aggregating platelets, which both stabilizes and gives mechanical strength to the somewhat fragile platelet plug.
Normal haemostasis is crucially dependent then on two factors:
- adequate numbers of normally functioning platelets and
- normally functioning blood clotting cascade.
In order to understand the defects of haemostasis associated with disease and how the results of the five blood tests are applied, it is helpful to examine platelets and the clotting cascade in a little detail.
Platelet production, structure and function
Like the other formed elements or cells that circulate in blood, platelets (alternative name thrombocytes) are derived from the stem cells of the bone marrow (see Chapter 15, Figure 15.1). A proportion of stem cells differentiate by stages within the bone marrow to megakaryocytes. Platelets are produced within the cytoplasm of these cells. Still within the bone marrow, platelets are released from mature megakaryocytes and pass from bone marrow to blood. Each megakaryocyte produces around 4000 platelets. Platelets have a lifespan of just ten days in blood so that constant bone marrow production is necessary. With a diameter of 1–2 mm, platelets are far smaller than either red cells or white cells. Like mature red cells, they have no nucleus.
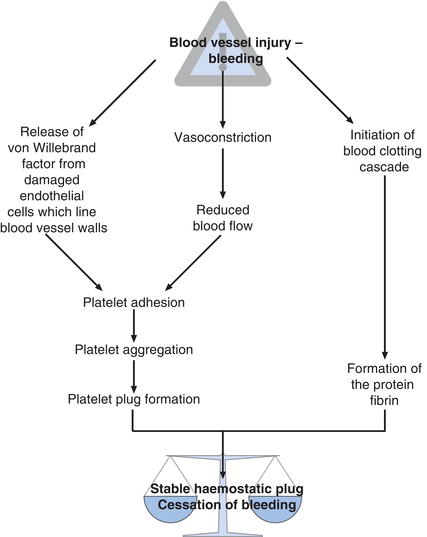
The principle function of platelets is to plug ‘holes’ in vessel walls caused during injury. The first stage in this process is adhesion of platelets to the wall of the damaged vessel. This adhesion is facilitated in part by a protein called von Willebrand factor, which is released from injured endothelial cells that line the internal surface of blood vessels. Adhesion proteins present on the surface of passing platelets bind to von Willebrand factor which itself is bound to proteins present on the surface of damaged endothelial cells. Following adhesion, platelets secrete many substances that modulate both the clotting cascade (explained further) and further platelet function. Among these are substances (e.g. ADP and thromboxane A2) which both induce platelets to stick to each other (aggregate) and swell in size. This process, called aggregation, continues until the mass of aggregated swollen platelets is sufficiently large to plug the damaged vessel.
Blood clotting cascade
As platelets are aggregating at the site of vessel injury, fibrin is being produced locally by the blood clotting (coagulation) cascade. This is a series of reactions in which specific proteins present in blood plasma, called clotting factors, are activated in sequence. Each activated factor promotes activation of the next and so on down the cascade; the final product being fibrin. These reactions are enzymic in nature. In their inactive state factors are pro-enzymes (i.e. have no enzymic activity). Enzymic action converts proenzymes (unactivated factors) to active enzymes (activated factors). Whilst most clotting factors are pro-enzymes/enzymes, some are not actually enzymes but substances that are required for some enzymic actions to occur. Thirteen clotting factors (Table 17.1) have been identified, numbered by convention in Roman numerals in the order in which they were first discovered. The once postulated Factor VI is no longer thought to exist.
The convention is that activated factors are distinguished from their inactive counterparts by the subscript ‘a’ and this is reflected in Figure 17.2, which is a simplified account of current understanding of the blood clotting cascade. The cascade comprises the intrinsic and extrinsic pathway, which both result in activation of Factor X. The route from activated Factor X to fibrin production is referred to as the common pathway.
Table 17.1 Blood clotting factors.
| Factor | Alternative name(s) | Notes |
Factor I | Fibrinogen | Precursor of fibrin – synthesised in the liver |
Factor II | Prothrombin | Pro-enzyme – |
Factor III | Tissue factor | Protein present in all tissues |
Factor IV | Calcium | Inorganic ion co-factor |
Factor V | Labile factor | Protein co-factor synthesised in the liver |
Factor VI | Once proposed but no longer thought to exist | |
Factor VII | Proconvertin | Pro-enzyme – |
Factor VIII | Antihaemophilic factor | Protein – co-factor |
Factor IX | Christmas factor | Pro-enzyme – |
Factor X | Stuart factor | Pro-enzyme – |
Factor XI | Plasma thromboplastin antecedent | Pro-enzyme |
Factor XII | Hageman factor | Pro-enzyme |
Factor XIII | Fibrin stabilising factor | Pro-enzyme |
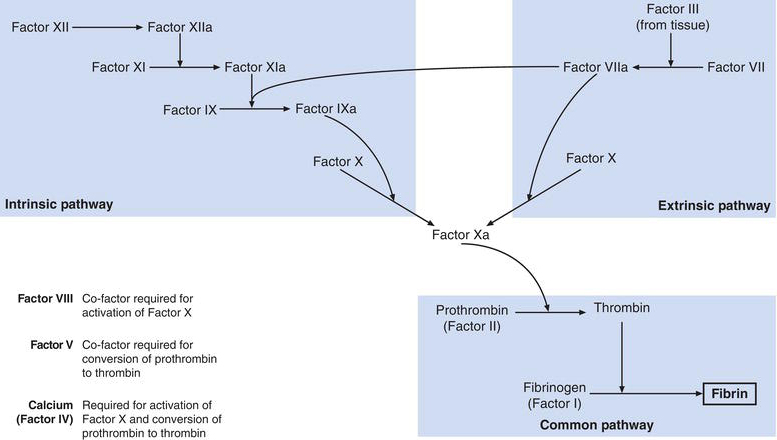
The intrinsic pathway is initiated when Factor XII is activated by contact with proteins within blood vessel endothelial cells, that are exposed as a result of vessel wall injury. Activated Factor XII then activates Factor XI, which in turn activates Factor X. Co-factors, Factor VIII (antihaemophilic factor) and Factor IV (calcium) are required for this last reaction.
The extrinsic pathway is initiated by Factor III. This is a substance called thromboplastin, found in most tissues and released to blood during tissue injury. Factor III activates Factor VII, which in turn activates Factor IX. In the final common pathway, activated Factor X activates Factor II (prothrombin) to the active thrombin, which in turn converts fibrinogen (Factor I) to fibrin. Factor V is a cofactor required for conversion of prothrombin to thrombin. The final formation of cross-linked fibrin polymers from fibrin (monomers) is dependent on activated Factor XIII.
Most of the factors of the blood clotting cascade including Factor I (fibrinogen), Factor II (Prothrombin), Factors V, VII, IX, X, XI and XII are synthesised in the liver and released to blood in their inactive form. The liver synthesis of Factors II (prothrombin), VII, IX and X is crucially dependent on vitamin K. Vitamin K is derived from two sources, diet and vitamin K synthesising bacteria normally present in the gut.
Formation of fibrin by the clotting cascade depends then on adequate plasma concentration of all clotting factors, which in turn is dependent on:
- Normally functioning liver (for their synthesis).
- Adequate dietary source of vitamin K.
- Normal bacterial flora in the gastrointestinal tract.
- Normal gastrointestinal absorption of dietary and non-dietary vitamin K.
Breakup of a blood clot – fibrinolysis
The fibrin-platelet ‘plug’ is a temporary structure produced in response to blood vessel injury to limit blood loss. It must eventually be degraded for healing to occur. A process called fibrinolysis is responsible for this degradation. Fibrinolysis also serves to limit fibrin clot growth so that it does not impede blood flow through the damaged blood vessel, and also limits the growth of tiny fibrin clots that are continuously being formed in patent undamaged vessels.
The fibrinolytic system thus keeps coagulation in check. It is useful to reflect that the fibrin clot is a dynamic structure in a state of growth or dissolution depending on whether coagulation or fibrinolysis is in ascendancy. When the clot is no longer needed because the injury is healing, or if a clot is beginning to form in an undamaged blood vessel, fibrinolysis should be in ascendancy.
Central to the process of fibrinolysis is a protein called plasminogen, which is synthesised in the liver and circulates in blood. Here it binds to fibrin, thereby being incorporated into a fibrin clot as it is being formed. This incorporation of plasminogen means that all blood clots contain the seeds of their destruction.
In common with factors of the clotting cascade, plasminogen is an inactive pro-enzyme so has no effect within the clot until it is converted in a controlled manner to the active enzyme, plasmin. On activation, plasmin enzymically cleaves the cross-linked fibrin polymers. The product of plasmin action is a heterogenous collection of cleaved fragments of fibrin that are swept in the blood stream from the currently dissolving fibrin clot. The cleaved fragments are collectively called fibrin degradation products (FDP), and among them is one kind of FDP called D-dimer, so called because it comprises two linked fragments (a dimer) from a part of the fibrin molecule called the D domain. Because they retain the cross linking that is peculiar to the structure of the fibrin within a fibrin clot, the presence of D-dimer in blood is objective evidence of current fibrin clot formation/degradation.
Laboratory measurement of platelet count, PT, APPT, TT and D-dimer
A platelet count is one of the tests included in a FBC. Laboratory measurement and sample requirements for FBC are discussed in Chapter 15. This section is concerned only with PT, APPT, TT and D-dimer.
Patient preparation
No particular patient preparation is necessary.
Timing of sample
Blood for PT, APPT, TT and D-dimer may be sampled at any time. However the proteins of the clotting cascade are not well preserved in a blood sample and falsely abnormal PT, APPT and TT results can occur if blood is not tested within four to six hours of sampling. This period can be extended to 15 hours if the sample is stored in a refrigerator at 4°C. Samples more than 15 hours old are not suitable for analysis.
Sample requirements
The tests are performed on plasma, the fluid that remains when all cellular elements are removed from anticoagulated blood. The blood must be collected into a tube containing the anticoagulant sodium citrate (usually a light blue top), which also preserves blood clotting proteins (factors). The volume required (usually 5 ml) is indicated on the bottle; it is important that neither more nor less than the volume indicated is added to the bottle. Gentle inversion to mix blood with the anticoagulant is essential. It is inadvisable to sample blood for these tests via an indwelling catheter, since heparin flushes are often used to keep these lines patent; falsely abnormal results will be obtained if blood samples are contaminated with heparin.
What do tests of blood coagulation measure?
Whilst it is clear that a platelet count is simply a measure of the number of platelets present in blood and the D-dimer test is a measure of the concentration of the protein D-dimer in blood plasma, it might be less obvious what is being measured by the three other tests considered in this chapter. Prothrombin time (PT), activated partial thromboplastin time (APPT) and thrombin time (TT) all test the ability of blood to generate fibrin by the blood clotting cascade. In essence, they all measure the time taken for a sample of patient’s blood plasma to form a fibrin clot in a test tube, after addition of a reagent that initiates the clotting cascade. Results are expressed in time (seconds). In the case of the prothrombin time test, commercially produced thromboplastin (Factor III) is added to the patient’s plasma. This is the clotting factor that initiates the extrinsic pathway, so that prothrombin time is a test specifically of the extrinsic and common pathways; a deficiency of any factor or factors of these two pathways (i.e. Factors X, VII, V, prothrombin and fibrinogen) will result in an abnormally prolonged time for a fibrin clot to form (i.e. PT will be raised).
In a similar way, by adding only an initiator of the intrinsic pathway to patient’s plasma, the APPT tests only the intrinsic and common pathways. In this case, an abnormally prolonged result indicates a deficiency of one or more of those factors required by the intrinsic and common pathways.
Finally, for the thrombin time test, thrombin is added to patient’s plasma. This is a test specifically of the final stages of the common pathway: fibrinogen to fibrin. An abnormally prolonged thrombin test indicates a deficiency of Factor I (fibrinogen).
So long as PT, APPT and TT are all normal, it can be assumed that there is a sufficiency of all coagulation (clotting) factors in the patient’s blood. As such, the normal results provide evidence that in the event of vessel injury the clotting cascade would work normally and perform its haemostatic function adequately.
Interpretation of results
Approximate reference ranges
Platelet count | 150–400 × 109/L |
Prothrombin time (PT) | 10–14 seconds |
Activated Partial Thromboplastin Time (APPT) | 30–40 seconds |
Thrombin Time (TT) | 14–16 seconds |
D-dimer | < 500 ng/ml |
[Caution: The D-dimer reference range varies greatly due to significant differences in methodology between laboratories.]
Critical values
Platelet Count | < 40 × 109/L or > 1000 × 109/L |
Prothrombin Time | > 30 seconds |
Activated Partial Thromboplastin Time | > 78 seconds |
Terms used in interpretation
Thrombocytes | alternative name for platelet. |
Thrombocytopaenia | reduced platelet count, that is < 150 × 109/L. |
Thrombocytosis | increased platelet count, that is > 400 × 109/L. |
Thrombosis | formation of a blood clot within a blood vessel. |
Haemophilia | a pathological state of decreased blood coagulability and therefore increased tendency to bleed. |
Thrombophilia | a pathological state of increased blood coagulability and therefore increased tendency to form blood clots (thrombi) inappropriately within patent blood vessels. |
Causes of decreased platelet count
Reduction in platelet numbers can be caused by:
- Reduced bone marrow production of platelets.
- Increased rate of platelet destruction.
- Increased rate of platelet consumption.
Reduced bone marrow production of platelets and resulting severe decrease in platelet numbers (may be less than 50 × 109/L) is a feature of aplastic anaemia, acute leukaemia, cytotoxic drug therapy and radiotherapy. Megaloblastic anaemia, that is anaemia invariably caused by deficiency of vitamins B12 or folate (Chapter 18), is also associated with decreased bone marrow production of platelets, although not usually severe. Secondary spread of primary cancer to the bone marrow can result in reduced platelet production so that a reduced platelet count is sometimes a feature of advanced cancer.
Immune thrombocytopaenic purpura (ITP) is the most common cause of increased platelet destruction. This relatively common disorder, which affects both adults and children but predominantly middle aged/elderly women, results from production of autoantibodies against a patient’s platelets. The cause of this autoantibody production is not known. In most cases the condition arises in otherwise well individuals, but it can be a secondary complication of some other primary disorder; these include systemic lupus erythematosus (SLE), infection with HIV and chronic lymphocytic leukaemia. Autoantibodies bind to platelets resulting in their premature removal and destruction within the reticuloendothelial system. Platelet lifespan is reduced from the normal ten days to just a few hours in this condition. Bone marrow production cannot keep pace with this rate of destruction and the reduction in platelet numbers is severe, in the range 10–50 × 109/L. A similar mechanism (antibody-mediated platelet destruction) is the cause of the reduced platelet count that sometimes complicates recovery from some viral infections (e.g. chicken pox and measles) in childhood.
Increased platelet consumption is a feature of disseminated intravascular coagulation (DIC), a not uncommon complication of many serious/critical conditions including: sepsis; severe tissue damage sustained during trauma or major surgery; obstetric emergencies such as pre-eclampsia/eclampsia and placental abruption; and incompatible blood transfusion. DIC is characterised by abnormal (inappropriate) activation of clotting cascade and platelet aggregation within blood vessels (i.e. thrombosis). The result is depletion of both platelets and clotting factors leaving these already very sick patients at risk of severe, even fatal, haemorrhage. The micro-thrombi formed during DIC may block blood vessels causing widespread ischaemic damage that may be sufficiently severe for progress to multi-organ failure.
The anticoagulant heparin and many other commonly used drugs can induce production of destructive antibodies directed at platelets with resulting increased platelet destruction. Drugs that may result in a reduction in platelet numbers include some anti-inflammatories, antibiotics (penicillin, suphonamide) and some diuretics (frusemide, acetazolamide).
Causes of increased platelet count
An increase in platelet numbers, termed thrombocytosis, is a feature of those malignant disorders of the bone marrow, collectively called the myeloproliferative disorders that are characterised by abnormal proliferation of myeloid stem cells. The myeloproliferative disorders in which an increase in platelet numbers can be expected include chronic myeloid leukaemia (around a third of all cases), polycythaemia vera (around a half of all cases) and essential thrombocythaemia (all cases).
A mild to moderate increase in platelet numbers (usually between 400 and 1000 × 109/L) is a relatively common phenomenon among acutely ill patients. The raised platelet count reflects bone marrow stimulation induced by blood loss, infection or tissue injury. It is termed secondary thrombocytosis to distinguish it from the thrombocytosis due to the primary bone marrow diseases already described. The conditions that might be associated with secondary thrombocytosis include: severe tissue injury due to major surgery or trauma; acute and chronic infection; malignancy; chronic inflammatory conditions such as rheumatoid arthritis or Crohn’s disease and chronic iron deficiency anaemia.
Surgical removal of the spleen (where platelets are normally sequestered when they are no longer viable), results in particularly high platelet count.
Consequences of abnormality in platelet numbers
Since platelets are required for normal haemostasis, patients with a reduced platelet count are at increased risk of excessive bleeding. Spontaneous bleeding may occur if platelet count falls below 50 × 109/L. Potentially fatal haemorrhage almost inevitably occurs if count falls below 5 × 109/L. An increased tendency to bleed due to platelet deficiency has several clinical manifestations, including increased menstrual blood loss (menorrhagia), easy or even spontaneous bruising, bleeding gums, nose bleeds (epistaxis), petechial (tiny pinpoint) haemorrhages into the skin giving a red rash like appearance and purpura. Widespread purple/brown discoloration of skin (ecchymosis), akin to bruising, occurs due to haemorrhage into tissues.
An increase in platelet numbers carries with it the theoretical risk of increased coagulability of blood (thrombophilia) and resulting thrombosis. In practice this risk of thrombosis is certainly not real until platelet count rises well in excess of 1000 × 109/L and in any case a direct link between severe thrombocytosis and increased risk of thrombosis is disputed.
Causes of increased PT, APPT and TT
An increase in any of these tests indicates a deficiency of one or more clotting factors. Deficiency may be congenital (i.e. inherited) or, much more commonly, acquired as a result of disease or anticoagulant drug therapy.
Haemophilia A, which accounts for around 85% of all the many known inherited blood clotting defects, is the most common inherited blood clotting deficiency. The defect is in the gene that codes for production of Factor VIII (also called antihaemophilia factor). This is the co-factor required for activation of Factor X by Factor IXa (Figure 17.2). The result of the genetic defect is a marked deficiency or complete absence of Factor VIII and severely impaired blood clotting. Without Factor VIII replacement therapy, affected patients are at risk of life-threatening haemorrhage. Chronic episodic bleeding into joints and resulting joint pain and deformity can lead to permanent disability for those not treated effectively. The extrinsic and common pathways are intact in the patient with haemophilia A so that PT and TT are normal. APPT (a test of the intrinsic pathway) however is abnormally prolonged. Haemophilia B (Christmas disease), a less common but equally devastating inherited defect of the clotting cascade in which the deficiency is of Factor IX rather than Factor VIII, also causes raised APPT.
Since many clotting factors are synthesised in the liver, multiple factor deficiency is a feature of liver disease (acute and chronic hepatitis, cirrhosis etc.). PT, APPT and TT may all be increased in severe liver disease, although a raised TT is less usual. Of the three, PT is the most sensitive marker of liver disease and is routinely used as a test of liver function both in its detection and to monitor progress.
The production of several factors of both the intrinsic and extrinsic pathway is dependent on vitamin K, so that deficiency of vitamin K is associated with an increase in both PT and APPT. Newborn babies are at risk of vitamin K deficiency, and thereby a bleeding abnormality called haemorrhagic disease of the newborn. This provides the rationale for the routine prophylactic administration of vitamin K to babies at the time of birth.
Adult vitamin K deficiency usually only arises as result of impaired absorption from the gastrointestinal tract. Absorption of the fat-soluble vitamin K is dependent on the bile salts contained in bile so that diseases that may be associated with poor absorption of vitamin K include those that result in obstruction of the bile tract (e.g. gall stones, cancer of the head of pancreas) and pancreatitis.
Dietary deficiency of vitamin K may occur in malnourished adults. Antibiotic use can be associated with deficiency because it affects the balance of normal gut flora, and therefore bacterial production of vitamin K. Whatever the cause, vitamin K deficiency is associated with an increase in PT and APPT; TT is normal.
Consumption and resulting deficiency of several clotting factors is a feature of the abnormal coagulation within blood vessels that characterises DIC (already discussed). PT, APPT and TT are all raised in DIC; the increase in TT is particularly marked.
Clotting factors are not well preserved in stored blood so that those patients who receive massive blood transfusion (i.e. total blood volume replaced) are paradoxically at increased risk of haemorrhage because the blood that is being transfused is relatively deficient of clotting factors. Increased PT, APPT and TT may all be evident in the patient who has received massive blood transfusion.
Consequences of increased PT, APPT AND TT
An increase in any or all of these tests implies a deficiency of one or more clotting factors, reduced blood coagulability and therefore a greater than normal risk of excessive bleeding.
Causes of increased D-dimer
Since, as already discussed, D-dimers are a product of thrombus degradation it is to be expected that markedly increased D-dimer concentration is a feature of thrombotic disease. Additionally, D-dimer is raised – often to a lesser extent – in a range of other diseases, in which for one reason or another, blood is in an abnormally pro-coagulant state. The list of common conditions associated with increased D-dimer is thus long (Table 17.2) and it is not difficult, after consideration of this list, to appreciate that the majority of hospitalised patients have increased D-dimer.
The D-dimer test has proven of greatest value in the assessment of patients suspected of suffering two common related thrombotic diseases: deep vein thrombosis (DVT) and pulmonary embolism (PE), which together are known as venous thromboembolism (VTE). Before addressing the way D-dimer is used in the diagnosis of VTE, we will consider the condition itself and the national co-ordinated strategy aimed at its prevention. Prevention of VTE is now considered an imperative of safe patient care that is led by specialist nurses and involves most hospital nurses1.
Venous thromboembolism (VTE)
Although thrombi (blood clots) can form in any part of the venous system, the most common site for clinically significant thrombi formation is in the deep veins that pass through the muscles of the legs. Typically, DVT originates in the veins of the calf muscles. The thrombus may be asymptomatic and resolve without clinical effect due to the physiological ‘clot busting’ action of the fibrinolytic system already described.
Alternatively, a DVT may persist and grow causing symptoms that include calf muscle pain and tenderness, along with swelling and reddening of the affected area. DVTs can extend proximally up the venous system to the knee, thigh and pelvis causing pain and so on at these sites. The larger the thrombus grows, and the further it extends, the greater is the risk of the potentially life-threatening consequence of DVT, PE.
Table 17.2 Non-exhaustive list of conditions that can be associated with increased D-dimer.
| Arterial thrombotic diseases |
Myocardial infarction (MI) |
PE occurs in patients with DVT when a thrombus or thrombus fragment breaks free and is swept away in the venous system to the right side of the heart, and onwards via the pulmonary artery to the vasculature of the lungs, where it eventually becomes lodged at some point in the pulmonary arterial tree, obstructing blood flow to a portion of lung. Technically, a thrombus or thrombus fragment that moves from its original site is called an embolus or more accurately a thromboembolus, and the process of movement through the vascular system is called thromboembolism.
The consequences of PE are variable and depend to a great extent on the site of the pulmonary vascular blockage, that is the area of lung that is deprived of blood by the blockage. Worst case scenario involves an embolus sufficiently large to block a major pulmonary artery with resulting extensive pulmonary infarction, cardiovascular collapse and sudden death before treatment (clot busting drugs) can be administered. This is rare (only around 5% of PE cases result in sudden death).
More commonly in patients with PE, a medium or small pulmonary vessel is blocked. PE signs and symptoms include: sudden onset of breathlessness, coughing up blood (haemoptysis), chest pain, cyanosis and hypotension. In any particular patient the presence and severity of these symptoms will broadly reflect the size of the area of lung that is inadequately perfused. Occlusion of very small pulmonary vessels may have no symptomatic effect, so that PE can be clinically silent.
Current understanding of the pathophysiology of VTE is based on the notion that thrombi form in veins for one or more of three reasons, collectively known as Virchow’s triad. They are:
- Reduced blood flow (slow blood flow promotes clot formation).
- Increased blood coagulability.
- Injury or disease mediated damage to the endothelium that lines the internal surface of blood vessels.
The relative importance of each of these three causative factors for development of VTE varies between individuals. Virchow’s triad helps to explain some of the many known risk factors that predispose to, or provoke, VTE (Table 17.3).
So, for example, one of the most widely publicised provoking triggers for VTE is long-haul air travel. This particular VTE risk factor arises from travellers having to sit for prolonged periods and the resulting immobilisation reducing blood flow up the veins of the leg. Blood clots (thrombi) are more likely to form in blood that is flowing sluggishly (first element of Virchow’s triad), so this explains why long-haul air travellers are at higher than normal risk of DVT. The risk is greater for those immobilised long-haul air travellers who are among the 1 in 20 of the population who also have an inherited defect (e.g. Factor V Leiden) that increases the coagulability of blood (second element of Virchow’s triad) or have recently suffered trauma or disease that has damaged the endothelium of blood vessels (third element of Virchow’s triad).
Given the long list of possible risk factors, the cause of VTE varies greatly between patients, but in one way or another Virchow’s triad is usually always satisfied.
VTE prevention
The extent of the health problem posed by VTE was highlighted in a House of Commons report published in 20052 which revealed that PE following DVT was responsible for close to 30 000 deaths in hospital each year in the UK, which represents 10% of all hospital deaths. With acknowledgement that treatment strategies directed at preventing VTE are highly effective, most of these deaths were judged to be preventable. The report sparked a nationally co-ordinated strategy for VTE prevention that is now being implemented.
Recently published national guidelines for VTE prevention3 recommend that all patients admitted to hospital be assessed for risk of VTE at the time of admission. All those judged to be at risk as a result of this assessment should be treated to prevent VTE. There are broadly two kinds of preventative treatments: mechanical and pharmaceutical. Mechanical VTE prophylaxis involves the use of compression stockings or similar device that promote blood flow through the veins of the leg (this is addressing the first of Virchow’s triad). Pharmaceutical prophylaxis – which addresses the second of Virchow’s triad – involves the use of an anticoagulant drug (heparin or heparin derivative) that artificially decreases the coagulability of blood. This is contraindicated for patients at high risk of bleeding, so all patients at risk of VTE should also have an assessment of bleeding risk before pharmaceutical prophylaxis is administered. The guidelines3 include the detail of how risk should be assessed, as well as the detailed application of both kinds of prophylactic treatment to specific patient groups, and other more general aspects of care that impact on VTE risk.
Table 17.3 Risk factors that predispose to, and provoke venous thromboembolism (VTE).
| Chronic Predisposing Factors |
| Inherited defects that increase blood coagulability Factor V (Leiden) This is an inherited defect in the gene that codes for production of clotting Factor V. The resulting abnormal Factor V is not effectively degraded so its action persists inappropriately, leading to excessive growth of fibrin clots. This defect is common, affecting 5% of the population and is significant in close to a third of all VTE cases. Factor II (G20210A mutation) This is an inherited defect in the gene that codes for production of Factor II (prothrombin). It results in excessive production of prothrombin and thereby excessive growth of fibrin clots. Affects around 2–3% of population Inherited deficiency of natural anticoagulant proteins The normal function of these blood borne proteins is to inhibit specific steps of the clotting cascade and thereby control fibrin clot formation. This control is lost in those with these individually rare inherited protein deficiencies. Acquired predisposing factors
Acute triggers – provoking factors
|
The use of D-dimer in the diagnosis of DVT/PE
D-dimer concentration is increased in patients with DVT and PE. Since it is also raised in many other conditions (Table 17.2) it is not possible to make a definitive diagnosis of DVT or PE on the basis of a positive (increased) D-dimer test. However so consistent is the finding of a positive D-dimer test in patients with DVT/PE that it is possible to reliably exclude DVT/PE if D-dimer test is negative, and this is the basis for the routine use of D-dimer test.
A definitive diagnosis of DVT/PE requires imaging evidence of a blood clot (thrombus) in a deep vein. Imaging is expensive, time consuming and ideally reserved only for those in whom there is very strong suspicion of DVT/PE. A negative D-dimer test in a patient with clinical details that are not highly suggestive of DVT/PE provides sufficient evidence to exclude the diagnosis without imaging. So the real value of the D-dimer test is to significantly reduce the number of patients that need be submitted for expensive imaging tests.
Monitoring anticoagulation therapy
One of the principle uses of the prothrombin time (PT) and activated partial thromboplastin time (APPT) tests is to monitor the anticoagulation therapy used in the treatment and prevention of VTE and other potential thromboembolic conditions.
The aim of anticoagulation therapy is to artificially decrease the coagulability of blood thereby preventing thrombi formation and reducing growth of existing thrombi. Heparin, administered intravenously or subcutaneously, is used for the treatment of existing DVT functions in this regard by inhibiting the action of several activated factors of the extrinsic pathway; it also impairs platelet function. To prevent further DVT episodes in a patient who has suffered DVT, long-term oral anticoagulant warfarin is prescribed. Warfarin operates by inhibiting the action of vitamin K in production of vitamin K dependent factors.
Of course reducing the coagulability of blood carries with it the risk of increased bleeding, so that anticoagulation therapy must be carefully monitored to ensure maximum level of anticoagulation consistent with minimum risk of excessive bleeding. Heparin is monitored using APPT and warfarin is monitored using the PT test. In the case of heparin use, dose is adjusted so that APPT is 1.5–2 times the normal value.
Warfarin, prothrombin and INR
As we have seen, when PT is used to investigate a suspected coagulation defect results are expressed in time (seconds). This is not the case when PT is used to monitor warfarin therapy. Instead, the international normalised ratio (INR) is used. The INR provides a way of expressing PT results to take account of the differing activity of commercially prepared tissue thromboplastin used in the test. This ensures that prothrombin results are more directly comparable and provides a more accurate control of warfarin therapy. The INR is defined as the patients PT in seconds, divided by the mean of the PT reference range, raised to the power of the international sensitivity index (ISI) of the particular thromboplastin being used:

Warfarin dose is adjusted so that the INR is maintained within a therapeutic range, which depends on the precise clinical reason for prescribing warfarin. For most patients the INR must be maintained within the range 2–3. In some circumstances increased level of anticoagulation is required in which case the therapeutic INR range is 3–4.5. If INR rises above the prescribed range (i.e. 2–3 or 3–4.5) dose must be adjusted downwards. All patients on long-term warfarin therapy should have their INR checked at regular (2–3 week) intervals.
Advances in technology have allowed the development of portable analysers for monitoring warfarin therapy. These allow anticoagulant monitoring outside the laboratory, either in clinics or primary care centres, where analysis is performed by nurses. In some anticoagulant clinics nursing staff are not only analysing patient samples but interpreting results and adjusting warfarin dose. The success of this arrangement has been demonstrated in several studies that have shown that a nurse specialist anticoagluant service is as effective as a haematology consultant run service, in maintaining therapeutic control among patients receiving warfarin4. Some studies5,6 have demonstrated the feasibility of patients themselves taking control of their own blood testing and dose management. Just as some diabetic patients monitor their own blood glucose and adjust insulin dose accordingly, so too some patients can successfully self-manage their long-term anticoagulation therapy, thereby avoiding the inconvenience of frequent hospital appointments.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


