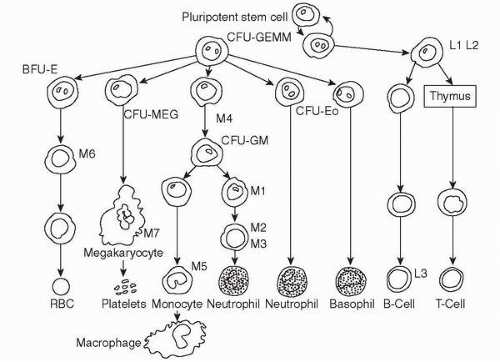
Leukemia Type (FAB classification) |
Defining Characteristics |
Prognosis |
|---|
Acute nonlymphocytic leukemia |
|
Cell maturation arrested along the myeloid cell line |
Prognosis of leukemia is highly dependent on the type of cytogenetic abnormality involved.
Cytogenetic abnormalities are divided into three groups: normal, favorable, and poor. |
Acute leukemia—minimally differentiated (M0 AML)
Acute leukemia—undifferentiated myelocytic (M1 AML)
Acute myelocytic leukemia AML (M2 AML) |
Age at onset: 18-25 years and 45-60 years, low to high WBC count at presentation. Splenomegaly, hepatomegaly, and lymphadenopathy are usually not seen. |
No abnormality, +8, 11q23, +21, del(9q) are normal t(8;21), inv(16) are favorable, and complex, −7, −5 are poor. M4 and M5 AML often involve MLL gene (11 gene) abnormalities, such as t(11;17)(q23;q21) or t(6;11)(q27;q23). 65% to 80% achieve remission with therapy, relapse is common. Median survival with treatment is 10-15 months. |
Acute myelomonocytic leukemia AM ML (M4 AML)
Acute monocytic leukemia AMoL (M5 AML) |
Often high WBC at presentation, with organ infiltration, such as gum hypertrophy, cutaneous leukemia, splenomegaly, hepatomegaly and lymphadenopathy. High incidence of central nervous system (CNS) involvement. |
Erythroleukemia EL (M6 AML) |
Complicated diagnosis, hard to distinguish from MDS, poor response to chemotherapy |
Poor prognosis |
Megakaryocytic leukemia (M7 AML) |
Extremely rare type of AML. Large organs (liver, spleen), accumulation of sclerotic tissue in bone marrow |
Poor prognosis |
Acute promyelocytic leukemia APL (M3 AML) |
Normal to moderately high WBC at presentation, high risk for DIC |
APL with t(5;17) with PML/RARα fusion protein is highly responsive to ATRA therapy and approximately 85% of patients achieve complete remission. |
Acute lymphocytic leukemia |
Erythroleukemia EL (M6 AML) |
Cell maturation arrested along the lymphoid cell line |
T-cell disease has a favorable prognosis |
Childhood acute lymphocytic leukemia (L1 ALL) |
Leukemia is most common form of cancer in children, and 75% of those are ALL. Age of onset: 2-3 years.
Splenomegaly, hepatomegaly, lymphadenopathy, CNS involvement are prevalent. |
Patient with t(9;22), t(1;19), infant with t(4;11), high WBC count, age <1 year or >10 years, CNS involvement, and longer time to achieve remission are associated with poor prognosis. |
Adult acute lymphocytic leukemia (L2 ALL) |
Philadelphia chromosome (t(9;22)) is the most common cytogenetics abnormality in adults. Splenomegaly, hepatomegaly, lymphadenopathy, CNS involvement are prevalent. |
Poorer prognosis than childhood ALL. Poor prognosis with t(9;22) involving 11q23 and t(1;19). Age >50 years, high WBC, longer time to achieve remission are associated with poorer prognosis. |
Burkitt’s type leukemia (L3 ALL) |
Age at onset: after 65 years, mature B-cell ALL, high CNS involvement and high tumor burden, high risk for TLS, rapid clinical course |
Worst prognosis in ALL, involve t(8;14)(q24;q11). High LDH associated with poor prognosis. |
Chronic myelocytic leukemia (CML) |
|
7% to 15% of adult leukemia, median age at onset 45-55 years, 90% of patients have + Philadelphia chromosome (t(9;22)), characterized by a chronic phase followed by an accelerating and blastic phase. |
Treatment is most effective in chronic phase. Accelerating and blastic phases are often refractory to treatment. 3 years of median survival with treatment. |
Chronic lymphocytic lymphoma (CLL) |
|
Age at onset: after 60 years, most common type of leukemia in adults, may be familial, prone to viral infection, patients with low-risk CLL often do not require treatment for many years. |
4-6 years of median survival rate. Low-risk patients mostly die of other causes, whereas the high-risk CLL patients die from disease-related complications within a few months of diagnosis. |