Bullous and Vesicular Diseases
Constance L. Hayes
OBJECTIVES
After studying this chapter, the reader will be able to:
Distinguish between the different disease processes of pemphigus vulgaris, bullous pemphigoid, epidermolysis bullosa, epidermolysis bullosa acquisita, and dermatitis herpetiformis.
Describe the tools available to make the diagnosis of pemphigus vulgaris, bullous pemphigoid, epidermolysis bullosa, epidermolysis bullosa acquisita, and dermatitis herpetiformis.
Define the therapeutic interventions used in treating pemphigus vulgaris, bullous pemphigoid, epidermolysis bullosa, epidermolysis bullosa acquisita, and dermatitis herpetiformis.
Identify the nursing interventions that assist in providing optimal care to patients.
Discuss the health maintenance important to patient/families with bullous/vesicular diseases.
KEY POINTS
Bullous and vesicular skin diseases can be acute but are primarily chronic blister-forming diseases. A genetic defect or immunological event causes a break in continuity within the epidermis or epidermal junction.
Topical treatments, drug therapies, and specific diagnostics are the cornerstone for health care management in bullous diseases.
Assessment for skin and systemic infections is imperative in all phases of care with impaired skin integrity.
Close monitoring of drug therapies for side effects and nutritional needs for growth/skin repair of the patient is essential.
Lesion biopsies, direct immunoflourescent studies, and blood studies are indicated for accurate diagnosis and treatment plans.
Pemphigus vulgaris is an autoimmune “intraepidermal” reaction against connections between epidermal cells. It has two distinct forms: P. vulgaris, the most common and severe with flaccid, fragile vesicles/bullae, which scab and heal without scaring, and P. foliaceus, less severe, superficial vesicles and crusted erosions that are absent in the mucosal areas.
Bullous pemphigoid is an autoimmune “subepidermal” reaction against epithelial cell attachments to the basement membrane and presents with subepidermal, intact, tense bullae that are less fragile and less severe than the group of ruptured, scabbed P. vulgaris bullae.
The “onion skin” approach describes a system to classify existing and newly identified genes and clinical phenotypes of inherited epidermal bullosa diseases.
Dermatitis herpetiformis is “virtually always” associated with celiac disease.
A multidisciplinary team of health care professions are needed to develop a workable comprehensive and optimal skin care plan for the patient and family.
PEMPHIGUS VULGARIS
I. OVERVIEW
A. Definition: Acute or chronic Pemphigus vulgaris (PV) is a rare, yet, serious bullous, autoimmune intradermal disease of the skin and mucous membranes caused by autoantibodies against the connections between cells in the epidermis.
B. Etiology
1. PV is due to autoimmune antibodies (IgG) against plakoglobin and desmogleins (desmoglein-1 and desmoglein-3—Dsg1, Dsg3), which are glycoproteins that help connect skin cells together. These are found in skin and mucous membrane keratinocytes at the spot desmosome location.
a. Dsg3 is more involved with mucous membrane disease.
2. Spot desmosomes, containing the desmogleins are found at the intracellular junctions in the stratum spinosum. In PV the desmogleins, Dsg1 and Dsg3, are not able to connect keratinocyte squamous cells together.
3. A genetic association has been proven through immunogenetics.
4. Additionally, various drugs may induce PV: antibiotics (penicillins, cephalosporin, rifampin), thiol drugs (captopril, enalapril, lisinopril, piroxicam, penicillamine), and pyrazolone derivatives (phenylbutazone, oxyphenbutazone).
C. Pathophysiology
1. Dsg1 and Dsg3 are glycoproteins in the desmosome of the keratocyte that “glue connecting skin cells” together. In PV, circulating IgG autoantibodies bind to the cell surface of keratinocytes in the lower part of the
epidermis and prevent Dsg3 specifically from providing its function of cell-to-cell adhesion.
epidermis and prevent Dsg3 specifically from providing its function of cell-to-cell adhesion.
2. These autoantibodies affect the spot desmosomes by disrupting and preventing connections between keratinocyte squamous cells of the epidermis, causing large, flaccid, superficial and fragile, intraepidermal bullae that rupture easily and scab.
3. Two variants of this disease are pemphigus vegetans (PVe) (a thickened verrucous-like change), which carries Dsg3 glycoprotein only, and pemphigus foliaceus (PF) (a more superficial variant), which carries Dsg1 glycoprotein only (Figure 20-1).
4. PV bullae are quite fragile and superficial as the roof consists of a thin portion of the upper epidermis that when dessicated leaves painful erosions with scabs that heal without scarring.
5. Painful oral erosions typically precede the skin blisters in PV by weeks or months.
6. PV is characterized by acute exacerbations and remissions, oftentimes requiring lifelong therapy. Severe cases can result in secondary infection and fluid loss.
7. Immunoflourescent biopsy indicates an intraepidermal pattern with a lacy outline of individual epidermal cells as autoantibodies bind to the junctions between the cells.
8. PF is less common and less severe with superficial vesicles that have crusted erosions, which are absent in the mucosal areas.
 FIGURE 20-1. Pemphigus foliaceus, preauricular lesions. (Image provided by Stedman’s.) |
D. Incidence
1. Usually occurs in 40- to 60-year-old persons; more common in people of Jewish, Mediterranean, and Indian decent
2. Equal incidence in males and in females
3. PV is the most common and most severe type.
E. Considerations across the lifespan
1. PV in pregnancy, otherwise known as pemphigoid gestationis (PG), is remarkably rare; yet, it is associated with increased risk of stillbirths, infant death, and intrauterine growth restriction (IUGR). As an autoimmune subepidermal blistering disease, there is a genetic susceptibility and hormonal influence from pregnancy, parturition, and oral contraceptives that are implicated in its etiology.
2. Neonatal PV may afflict up to 2% to 4% of infants born to mothers with PV; it is thought to occur from passive transplacental transfer of maternal antidesmoglein IgG antibodies to the fetus, which resolves with a few weeks of birth.
II. ASSESSMENT
A. History and physical examination
1. Vesicles and bullae are flaccid, easily ruptured, and weeping and arise on normal or erythematous skin. Subsequent erosions can bleed easily. Crusts may form and can affect any skin surface including the scalp.
2. The arrangement is randomly scattered, but there is a high propensity for oral mucosa; esophageal or vulvar involvement may occur.
3. Nikolsky sign is positive: Dislodging of the epidermis occurs with the application of lateral finger pressure where a vesicle is located, which leads to an extension of the vesicle and/or removal of epidermis (erosion). (This test is no longer used since it is nonspecific and because it creates an additional lesion.)
B. Specific skin changes
1. PV manifests with superficial erosions and flaccid weeping blistering or bullous lesions, 1 to 3 cm, that rupture easily, leaving denuded areas that crust.
2. Bullae may form from normal-looking skin or an erythematous base, with a predilection for seborrheic skin areas—face, midchest, and upper back.
3. Mucous membranes may involve mouth, vaginal, perianal, and conjunctiva. Site of first lesions may be oral mucosa, occurring in 50% to 70% with PV (Figure 20-2).
4. Symptoms of burning and/or pain occur in affected areas. Conjunctival involvement, which is rare, manifests with photophobia, irritation, and pain.
5. Patients may experience weakness, malaise, and weight loss especially if there is prolonged oral mucosal involvement.
C. Differential diagnosis
1. Include most bullous presentations: bullous pemphigoid, epidermolysis bullosa acquisita (EBA), dermatitis herpetiformis (DH), erythema multiforme, Grover
disease, Hailey-Hailey disease, Darier disease, pemphigus erythematous, and pemphigus foliaceus.
disease, Hailey-Hailey disease, Darier disease, pemphigus erythematous, and pemphigus foliaceus.
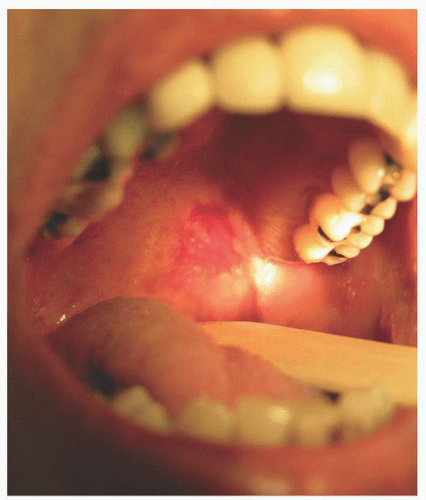 FIGURE 20-2. Pemphigus vulgaris. The first lesions may be in the oral mucosa. (From Dr. Barankin Dermatology Collection.) |
2. Similar immune disorders: linear IgA dermatosis and IgA pemphigus.
D. Diagnostic tests
1. Biopsy the edge of an early small bulla or, if not present, the margin of a larger bulla or erosion for histopathological light microscopy. There is a loss of intercellular cohesion (acantholysis) in the desmosomes of the keratinocytes near the bottom of the epidermis.
2. Direct immunofluorescence (DIF) testing is performed on a perilesional skin biopsy to detect IgG antibodies against Dsg1 and Dsg3; a lace-like pattern outlining individual cells develops where autoantibodies bind at the junctions between cells.
3. Enzyme-linked immunosorbent assay (ELISA) can help detect autoantibodies to Dsg1 and Dsg3 and is helpful to direct therapy (medication dosing) as it usually correlates with activity of the disease.
4. Complete blood counts (CBC) for leukocytosis, albumin, and electrolyte panels for disease management of potential infections, electrolyte, and protein losses/imbalance.
5. Prior to initiating corticosteroid or immunosuppressive therapies, secure chest x-ray, serum glucose, tuberculin skin test, and CBC.
6. In PV, significant esophageal symptoms or even strictures may occur, which would necessitate endoscopy and/or biopsy.
III. COMMON THERAPEUTIC MODALITIES
A. Systemic therapy
1. Systemic corticosteroids for blister suppression are the principal method of treatment with oral starting doses of 0.5 to 1.0 mg/kg. The dose may be doubled until remission is achieved.
2. Intravenous immunoglobulin (IVIG) has been used as second-line therapy where oral medication approaches were ineffective, are contraindicated (as in pregnancy), or had caused significant side effects.
3. Azathioprine immuno-suppressant 1 to 3 mg/kg/d, immunosuppressant may help during pregnancy when trying to minimize systemic steroid therapy for severe cases.
B. Local or topical therapy
1. Topical corticosteroids (both as creams or ointments on the skin and gel or liquid for mucosal lesions) can be beneficial for pruritic and erythematous areas.
2. Intralesional steroids may be used for resistant localized lesions.
3. Topical antibiotic creams or ointments such as silver sulfadiazine or mupirocin for blisters or erosions.
4. Medicated baths (e.g., potassium permanganate [KMNO4] or oilated oatmeal) and/or wet dressings (aluminum subacetate, KMNO4, or Domeboro solution) are useful for their antibacterial and cleansing effects, change every 2 hours.
C. Medical intervention
1. Primary goal for treatments: Reduce autoantibody synthesis by immune system.
2. Immediate start of high-dose systemic corticosteroids with tapering after good clinical response over several months. Adding immunomodulators or antimetabolites can help prevent significant steroid side effects.
3. In widespread disease, electrolytes of sodium, chloride, calcium may decrease while potassium increases. Secure electrolyte panel.
4. Patients with significant impetiginization of the eroded areas may show an increased WBC count. Secure CBC and check platelets with certain drug therapies.
5. Patients treated with systemic corticosteroids or biological immunomodulators should be placed on calcium and vitamin D, screened annually for tuberculosis/HIV, have comprehensive blood work and blood pressures every six months for close monitoring.
6. Patients with severe cases may require hospitalization on an inpatient dermatology unit or burn unit. Plasmapheresis can be used in conjunction with corticosteroids for more rapid control in extremely severe cases.
D. Medication—drug therapy
1. Immunosuppressants such as methotrexate, 20 to 50 mg per week; cyclophosphamide orally 1 to 3 mg/kg/d; or cyclosporine are generally used for remission induction or as an adjunct to steroid therapy.
2. Biologic immune response modifiers, like rituximab or mycophenolate mofetil, help reduce autoantibody synthesis and can be added to systemic steroid therapy.
3. Alternatives to immunosuppressives include diaminodiphenylsulfone, tetracycline, or minocycline, which are used alone or in combination with high-dose steroids.
4. Less frequently, antimalarial drugs such as chloroquine, hydroxychloroquine, or a combination of both are used.
E. Diet management
1. Support nutritious diet to replace protein and fluid losses from erosions. Full liquid or mechanical soft diets in acute phases with oral lesions are helpful.
2. High-protein liquid nutritional supplements are easily ingested until ulcers heal.
3. Provide topical anesthetic (viscous lidocaine) as an oral rinse prior to meals, in 4.
4. Use sponge swabs for oral hygiene when soft bristle toothbrushes are not tolerated.
F. Surgical interventions. None noted.
G. Supportive skin care
1. Continuous wet dressings, changed every 2 to 3 hours, prevent the dressing from drying/adhering to the skin and are preferred over “wet-to-dry” dressings.
2. Apply creams or ointments to dressings rather than eroded skin to reduce pain when changing dressings; may switch to body temperature lotions or dusting powder if creams or ointments are not tolerated
3. Refrain from using any tape that may exacerbate bullae formation.
4. Maintain body temperature with warm wet dressings, wrap quickly with towels.
H. Other nursing interventions
1. Explain need to monitor progress on latent infection reactivation or current infection exacerbation.
2. Assess patient/family’s financial ability to provide balanced diet and supplements and proper skin care supplies with dressings and creams.
3. Assess patient/family’s understanding and motivation to perform recommended treatments with available home bathing facilities.
I. Complimentary alternative medicine (consult primary provider before use)
1. Swedish bitters liquid may relieve tongue blisters by diluting 1 tablespoon in a glass of water or herbal tea swish/gargle prior to eating and as needed.
2. Soap wart (natural plant, saponaria officinalis): Cleanse blistered skin with soap wart solution of 1 teaspoon soap wart to 1 cup tepid water; drying naturally helps eliminate toxins.
IV. HOME CARE CONSIDERATIONS
A. Continuity of care concerns
1. Develop an achievable care plan with the patient/family for regular follow-ups and emphasize need to comply with all provisions of the treatment regimen.
2. Identify if the patient is able to care for self or identify a caregiver.
3. Call patients routinely during the acute phase to assess and prevent complications.
4. Provide contact numbers of provider(s) for patient inquiries and a 24-hour telephone number for emergencies.
Pemphigus Vulgaris and Bullous Pemphigoid
Teach proper aseptic skin care techniques with patient/family members to avoid potential skin infections and allow for return demonstration opportunities.
Explain the need to avoid individuals with infectious processes (flu, colds, coughs, skin infections, like herpes simplex lesions) especially since most therapies involve some degree of immunosuppression.
Review side effects of corticosteroid therapies (GI bleed, weight change, mood swings, acne, bone density changes, skin changes with overuse, and steroid-induced diabetes).
Reinforce need to follow up with providers to assess treatment outcomes and prevent infections.
BULLOUS PEMPHIGOID
I. OVERVIEW
A. Definition. Bullous pemphigoid (BP) is an uncommon, autoimmune, subepidermal blistering disease that mainly afflicts the elderly.
B. Etiology
1. BP is due to antibodies directed against the basement membrane zone of the epidermis (the “foundation” of the skin), creating subepidermal, mostly intact and less fragile, tense bullae than PV.
2. Drug reactions may induce BP: furosemide, penicillin, captopril, B-blockers, sulfonamides, terbinafine, penicillamine, NSAIDs, and potassium iodide.
C. Pathophysiology
1. Autoantibodies in BP, IgG, and, in some cases, IgA mediate tissue-destructive proteases, BPA1 and BPA2, against “hemidesmosomes” (intracellular junctions which attach epithelial cells to the dermis basement membrane).
2. BP can be self-limiting and may go into remission 2 to 6 years after onset.
3. Immunoflourescent findings include a line at the BSE of the epidermis (not the lace-like pattern outlining epidermal cells as seen in PV).
4. Variants of BP are:
a. Localized—usually limited to lower extremities and responds well to treatment; may progress to generalized disease.
b. Benign mucosal pemphigoid—synonym for cicatricial pemphigoid or scarring pemphigoid; recurring bullae either on a mucous membrane or on the skin near an orifice; and has the tendency to scar.
D. Incidence
1. Rarely seen in children, BP usually presents in adults over the age of 60 to 70 years.
2. Estimated incidence is 7 to 14 per 100,000 per year, afflicting men more than women by 2:1 in some studies.
II. ASSESSMENT
A. History and physical examination
1. Presents as a chronic blistering eruption on an inflammatory or urticarial base.
2. Manifests with tense, intact, serum-filled bullae on normal and erythematous skin that rupture easily—but less fragile than PV bullae.
3. Urticarial plaques and papules can precede bullae formation by months.
4. The distribution is generalized or localized; small oral ulcers occur infrequently.
5. The sites of predilection are generally flexural and include axillae, medial aspects of thighs and groin, flexor aspects of forearms, and lower legs.
6. Remissions in most adults occur within 5 years, more rapidly in children.
B. Specific skin/nail changes (Figure 20-3)
1. Begins as a hive-like preblistering rash where pruritus and nonspecific eczematous or papular lesions can precede the bullae.
2. Pruritic fixed urticarial plaques and tense bullae can be seen together; when the latter rupture, erosions can occur.
3. Oral lesions involved in 10% to 35%, much less common than PV.
4. Nail dystrophy associated with BV includes hyperkeratosis, shedding, hemorrhage, and horizontal ridging.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


